
Abstract
Obligate parthenogenesis is found in only 0.1% of the vertebrate species, is thought to be relatively short lived and is typically of hybrid origin. However, neither the evolutionary persistence of asexuality in vertebrates, nor the conditions that allow the generation of new parthenogenetic lineages are currently well understood. It has been proposed that vertebrate parthenogenetic lineages arise from hybridisation between two divergent taxa within a specific range of phylogenetic distances (the ‘Balance Hypothesis’). Moreover, parthenogenetic species often maintain a certain level of hybridisation with their closest sexual relatives, potentially generating new polyploid hybrid lineages. Here we address the role of hybridisation in the origin and evolutionary lifespan of vertebrate parthenogens. We use a set of microsatellite markers to characterise the origins of parthenogens in the lizard genus Darevskia, to study the distinctiveness of sexual and asexual taxa currently in sympatry, and to analyse the evolutionary consequences of interspecific hybridisation between asexual females and sexual males. We find that parthenogens result from multiple past hybridisation events between species from specific lineages over a range of phylogenetic distances. This suggests that the Balance Hypothesis needs to allow for lineage-specific effects, as envisaged in the Phylogenetic Constraint Hypothesis. Our results show recurrent backcrossing between sexual and parthenogenic Darevskia but neither gene flow nor formation of new asexual lineages. We suggest that, along with their demographic advantage, parthenogens gain additional leverage to outcompete sexuals in nature when the retention of sexual reproductive machinery allows backcrossing with their sexual ancestors.
Similar content being viewed by others


Introduction
Asexual reproduction is distributed across all major clades of the tree of life. Exclusively asexual species are expected to be short lived (Otto 2009), typically have a ‘twiggy’ phylogenetic distribution (Bell 1982) and originate from sexual ancestors (Avise 2008). Many studies have focused on the evolution of sex, trying to understand how such a costly reproductive mode is so successful in nature (Weismann 1889; Maynard Smith 1978; Otto 2009). This paradox of sex has been tentatively explained by the superior potential for adaptation to changing environments of sexual species compared with asexual ones (McDonald et al. 2016; Luijckx et al. 2017). Given the putative young age of (most) asexual species, we can deduce that sexual species constantly have the potential to give rise to new asexual lineages, but the predicted offset between generation and extinction of asexual lineages has not often been studied empirically.
The maintenance of asexual species (which constitute clusters of lineages of similar genotype and phenotype; cf. Barraclough 2010) depends on the generation of asexual lineages from sexual progenitors and their loss, either through accumulation of deleterious mutations (Haigh 1978), failure to adapt (Lively 2010) or neutral processes (Schwander and Crespi 2009; Janko 2014). Asexual invertebrates generally result from spontaneous thelytokous parthenogenesis (Bullini 1994). Parthenogenetic vertebrates are less common than invertebrate asexuals (Beukeboom and Vrijenhoek 1998), they are obligate, mostly recent (Beukeboom and Vrijenhoek 1998; Avise 2008) and often include polyploids (Bullini 1994). Parthenogenetic vertebrates typically originate through hybridisation between two individuals of different taxa (e.g. Choleva et al. 2012; Lutes et al. 2011; but see Sinclair et al. 2010), often resulting in sexual–asexual complexes including a network of species that recurrently hybridise (Danielyan et al. 2008), and eventually allow for the origin of new parthenogenetic species (Taylor et al. 2015). Cases where gene flow occurs between proposed evolutionary units, or where there is a recurrent origin of new asexual lineages from sexual parents, question the current definition and applicability of some species concepts (Coyne et al. 1988; Birky and Barraclough 2009). However, repeated hybridisation events often underlie the origin of parthenogenetic vertebrate species (as in Aspidoscelis sp. (Reeder et al. 2002), Lepidodactylus lugubris (Trifonov et al. 2015) and Cobitis elongatoides-taenia (Choleva et al. 2012)). Since these events result in phenotypically and genetically similar lineages, they are consistent with the definition of asexual species used here.
Two general hypotheses have been put forward regarding the conditions required for interspecific hybridisation to give rise to parthenogenetic lineages: the Balance Hypothesis (BH) (Wetherington et al. 1987; Moritz et al. 1989; an idea first discussed with reference to plants by Ernst 1918 (cited in Bartos et al. 2019)) and the Phylogenetic Constraint Hypothesis (PCH) (Darevsky 1967; Murphy 2000). The BH proposes that parthenogenetic vertebrates arise by hybridisation between two sexual species divergent enough to disrupt meiosis in the hybrids, yet not so divergent as to compromise hybrid viability or (parthenogenetic) fertility (Moritz et al. 1989; Kearney et al. 2009). Under the BH, pairwise distances between parental species, at the inferred time of origin of the parthenogenetic hybrids, should fall within an interval that is constrained at both ends. Before that interval, viable and sexually fertile offspring are formed, and after that window hybrids are either inviable or both sexually and parthenogenetically infertile. This window is likely to be much shorter than the time since the separation of the parental lineages. Any pair of clades that has passed through this interval will have had the opportunity to generate parthenogenetic lineages. We consider this the core of the BH. Moritz et al. (1989) further suggested that ploidy changes might alter the trade-off between incompatibility and disruption of meiosis, and so the window for the origin of parthenogenetic lineages. We view this as an extension of the core BH. The PCH proposes that asexual lineages originate from hybridisation between the pairs of sexual species that, alone or in combination, possess lineage-dependent genetic factors that allow them to interbreed and produce viable hybrids capable of reproducing parthenogenetically. One lineage may possess particular factors that need to come from the maternal lineage, for example, associated with female meiosis or egg formation. In this case, the PCH also predicts that hybridisation events generating parthenogens will be directional, with species from different clades contributing either the maternal or paternal ancestry (Murphy 2000). These two hypotheses are not mutually exclusive. Under the PCH, hybridisation between very close or very distant lineages is unlikely to generate parthenogenesis. The BH does not exclude a higher probability of generating parthenogenetic lineages from some combinations of parental species than others. Nevertheless, it is helpful to consider whether the origin of parthenogenetic lineages is mainly constrained by lineage-specific effects (PCH) or by genetic distance (BH) because this might give insight into the phylogenetic distribution of asexual vertebrates and the conditions needed for their generation and persistence.
The model system used in this study, Darevskia lizards, has a hybridisation-rich evolutionary history, and therefore is a suitable model to study the correlation between hybridisation and parthenogenesis. All of its asexual lineages are reported to be of hybrid origin (Murphy 2000; Freitas et al. 2016), there is evidence for recurring mating between asexual females and sexual males when in sympatry, generating polyploid backcrosses (Darevsky and Danielyan 1968; Danielyan et al. 2008) and for frequent interspecific hybridisation between sexual species (Darevsky 1967). Given that maternal and paternal ancestors of parthenogenetic Darevskia are reported to be limited to a few sexual species, this model appears to fulfil the predictions of the PCH (Darevsky 1967) although this observation alone does not exclude the BH. However, inferences of the origin of the different parthenogenetic species and the identification of the polyploids have been based on limited evidence, and require genetic confirmation.
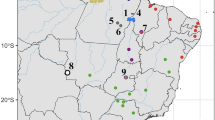
Initial estimates of the phylogenetic relationships of sexual species (Murphy et al. 1996; Murphy 2000) suggest Darevskia is divided into three main clades (Fu et al. 1997). Considering the asexual species and their putative parents (Fig. 1), D. armeniaca is thought to have resulted from hybridisation between D. mixta and D. valentini (Fu et al. 2000a, Moritz et al. 1992), while both D. unisexualis and D. uzzelli are thought to have resulted from hybridisation between D. raddei and D. valentini (Fu et al. 2000b). The maternal species was always D. raddei or D. mixta (placed in the same unresolved terminal clade by Murphy 2000) and the paternal species was D. valentini or D. portschinskii (the latter, and its descending asexual hybrid lineages, were not analysed in this study). Darevskia rudis is the third species from the rudis clade, phylogenetically very close to D. valentini and D. portschinskii, and there is evidence for between-species gene flow within this clade (Tarkhnishvili et al. 2013). The relevance of D. rudis to the origin of the parthenogens is still unclear. Hybridisation between parthenogenetic Darevskia females and males of their sexual parental species has been reported, based on morphology and karyology of a limited set of individuals (Danielyan et al. 2008). Reported triploid hybrids (3n = 57) included both females and males, with unknown fertility and varying levels of reproductive organ development (Danielyan et al. 2008). Since eggs of different ploidy may develop in the same oviduct, it is not clear if hybrids between sexual males and asexual females are always polyploid or can also be diploid.

Previously inferred relationships between sexual species D. mixta (mix), D. valentini (val), D. rudis (rud) and D. raddei (rad) and their hybrid parthenogenetic descendant lineages, D. armeniaca (arm), D. unisexualis (uni) and D. uzzelli (uzz)
The goal of this study was to address three questions related to the origin of parthenogenesis in vertebrates, using Darevskia as a model: (1) Is the origin of hybrid asexuality in Darevskia more consistent with the BH or with the PCH? (2) Did multiple hybridisation events originate each recognised parthenogenetic Darevskia species? (3) Is there ongoing backcrossing with gene flow between the parthenogens and their sexual parents?
To address these issues, we used microsatellite markers and genotyped individuals from a wide range of localities from Armenia, Turkey, Georgia and Iran. We reassess the inferred parentage of three parthenogenetic Darevskia: D. unisexualis, D. uzzelli and D. armeniaca, comparing matrilineal (mtDNA) and nuclear (maternal + paternal) lineages of the putative parents and parthenogens. Current sympatric localities were surveyed, and the evolutionary significance of hybrids is discussed. Our results indicate that the origin of parthenogenetic Darevskia is more consistent with PCH than with BH. We found no evidence that recurrent backcrossing between sexual and parthenogenetic Darevskia has led to gene flow. Finally, our results suggest that, while they retain sexual reproductive machinery, parthenogenetic vertebrates impose an additional cost on sympatric sexual populations by backcrossing with their sexual parents.
Material and methods
Sample collection
In this study, 378 Darevskia individuals were analysed (Table S1, Fig. S1). Samples were collected between 2007 and 2011 in Armenia, Turkey, Georgia and Iran, aiming to include representatives of each species’ distribution range. Seven species were included in the analyses, three parthenogenetic (D. armeniaca, D. unisexualis and D. uzzelli) and four sexual (D. mixta, D. raddei, D. valentini and D. rudis). Individuals were provisionally identified in the field based on overall morphology, size, colour pattern and scutellation (Arakelyan et al. 2011).
Four sympatric localities in Armenia where sexual and parthenogenetic species coexist were sampled (Table S2). In Kuchak, previous studies have reported backcross individuals between parthenogens and sexual species (Danielyan et al. 2008). Three species have been found: two parthenogenetic (D. armeniaca and D. unisexualis) and one sexual (D. valentini), the putative paternal species for both parthenogens present. Putative backcross individuals, D. armeniaca × D. valentini and D. unisexualis × D. valentini, have been identified based on morphology, and polyploidy of some individuals has been confirmed by karyology, identifying both triploids and tetraploids (Danielyan et al. 2008). In total, 150 individuals were sampled, together with individual information (location, pictures, temperature) reported elsewhere (Carretero et al. 2018; Sillero et al. 2018). In Sotk, the sexual D. valentini and the parthenogen D. armeniaca have been found in sympatry, and individuals morphologically similar to D. armeniaca × D. valentini backcrosses were tentatively identified based on large body size and intermediate colouration. In the remaining two localities, Lchap and Lchashen, the sexual D. raddei was found together with its hybrid descendant, the parthenogen D. unisexualis (Lchap), or with the sexual D. valentini (Lchashen).
Genotyping
Genomic DNA was extracted from 30 mg of tail-tip tissue following standard high-salt protocols (Sambrook and Russell 2001). From a total of 74 tested microsatellite loci developed previously for other lacertid lizards, 12 polymorphic markers were selected on the basis of reliable amplification and heterozygosity: D119, C118, C113 (Remón et al. 2008), Pb55 (Pinho et al. 2004), Lv-4-72 (Boudjemadi et al. 1999), P011, P054 (Wellenreuther et al. 2009), Ph39, Ph124, Ph128, Ph170 (NCBI accession numbers: KC869962, KC869964, KC869956, KC869961) and Du323, Du47, Du418 (Korchagin et al. 2007) (for more information see Table S3).
PCR amplifications were carried out using the Multiplex PCR Kit (QIAGEN) following the manufacturer’s instructions in a final 10 μl volume, including a negative control. Amplicons were separated by size on an ABI3130xl Genetic Analyzer. Allele sizes were scored against the GeneScan500 LIZ Size Standard using GENEMAPPER 4.0 (Applied Biosystems) and manually checked twice, independently. To control against allelic dropout, which is expected to be higher in polyploids due to the greater number of amplicons in each PCR reaction, 35–45% of genotypes per marker were repeated, including all the putative polyploids, in independent PCR reactions (Table S3). For the repeated samples, loci were genotyped individually to confirm that the third (or fourth) allele scored was not an artefact of interaction between the different primer pairs in the multiplex.
To test for the presence of null alleles, genotyping errors and allelic dropout, we used Microchecker 2.2.3 (Van Oosterhout et al. 2004). These tests were performed only on D. valentini (excluding individuals from sympatric localities) because it was the species with most individuals, and the assumptions of this analysis are not appropriate for parthenogenetic species or for mixed-species samples. Tests for Hardy–Weinberg equilibrium (HWE) and linkage disequilibrium (LD), and standard genetic diversity measures, observed (HO) and expected (HE) heterozygosities, allele frequencies and allelic richness, were obtained using Cervus/Fstat v2.9.3.2 (Goudet 1995). The critical probability for each test was adjusted with a sequential Bonferroni correction (Rice 1989). Only markers in HWE and without significant LD were used hereafter.
Sympatric localities: ploidy determination
Individuals found at the sympatric localities were analysed for their ploidy and parentage, testing the outcomes of hybridisation between parthenogenetic females and sexual males. Ploidy level was identified as the maximum number of alleles found among the markers used (ploidy function in Polysat; (Clark and Jasieniuk 2011)). All individuals characterised as triploids had at least two markers with three different alleles. Given the multiple ploidy levels in the sympatric localities (see “Results” section), traditional diversity measures and population genetic analyses could not be applied to these datasets. Instead, we calculated inter-individual genetic distances using the Bruvo method (Bruvo et al. 2004) implemented in Polysat (detailed information in SI).
Sympatric localities: principal coordinate analysis
Principal Coordinate Analyses (PCoA) were performed on the individuals from the sympatric localities, and then plotted against the corresponding species from elsewhere in the distribution. Bruvo distances were used as a dissimilarity matrix, and sympatric localities were grouped in pairs according to their species makeup (Table S2). Kuchak and Sotk constituted the first pair, both with D. valentini (sexual paternal species) and parthenogenetic species, and Lchap and Lchashen the second, both with D. raddei (sexual maternal species) and others.
Cluster analyses
Cluster analyses were performed on the dataset of all diploid individuals, using a hierarchical approach. First, we determined the optimal number of clusters (K) on the dataset of all diploid individuals, excluding the diploids from the sympatric localities. K was determined using the find.clusters option in the ADEGENET package (Jombart et al. 2010) by comparing the different clustering solutions using a Bayesian Information Criterion (BIC). For similar BIC values, the optimal K value was selected based on concordance between the clusters and described taxa. This value of K was then compared with ∆K and the rate of change of the log probability of the data between successive K values calculated from structure analyses on the same dataset (see below).
Second, we visualised the relationships among sexual and parthenogenetic species using the diploid individual dataset, now including the diploids from the sympatric localities. We performed an exploratory Discriminant Analysis of Principal Components (DAPC) also implemented in ADEGENET (Jombart et al. 2010). We used the K value calculated previously as the number of clusters to which individuals were assigned, and DAPC was used to ordinate individuals according to axes that maximise cluster distances relative to variation within clusters. We also calculated the pairwise FST values between the species (diploids from the sympatric localities were separated into independent groups) in this same dataset with pairwise.fst in the package hierfstat, version 0.04–22 (Goudet 1995). To avoid effects on FST due to small sample size, the pairwise FST was estimated only when seven or more individuals were available per species and population.
Third, we determined the numbers of (diploid) parthenogenetic lineages and hybrid origins of the parthenogenetic diploid individuals (including diploid parthenogens from the sympatric localities) by conducting assignments using DAPC membership probability values for each parthenogenetic species alone. To further confirm the number of hybrid origins per species, we calculated the Bruvo (Bruvo et al. 2004) pairwise distance distribution among individuals of each parthenogenetic species. The Bruvo method was used here so that comparisons with polyploids were made possible. Lynch distances showed a similar arrangement of clusters and distances between species and hybrids (Fig. S2, more information in the supplementary material). Hartigan’s dip test (implemented in R package diptest version 0.75–7: https://CRAN.R-project.org/package=diptest) was used to assess departure from an unimodal distance distribution.
Fourth, to test whether genetic structure was due to isolation by distance (IBD) or the presence of distinct groups within taxa, IBD analysis was performed on the nominate sexual species D. raddei, D. valentini and D. mixta (including sympatric localities), with the ADEGENET package in R (Jombart et al. 2010).
Last, population structure and the ancestry of the parthenogens were investigated using the Bayesian multilocus clustering analysis implemented in STRUCTURE v2.3.4 (Pritchard et al. 2000; Hubisz et al. 2009) (for run details see SI). The sexual species (diploid individuals from sympatric localities included) were used as ‘learning samples’ (PopFlag = 1) (Murgia et al. 2006) to define the cluster membership when, subsequently, diploid parthenogenetic individuals were included in the dataset. Cluster membership was assigned according to the optimal number of clusters calculated previously (K), and excluding the clusters of parthenogens (since only sexual species were used as ‘learning samples’). Given their hybrid origin, parthenogens are expected to have half of their ancestry from the maternal species and the other half from the paternal species, with little variation due to their clonal reproduction. The ancestry of the parthenogenetic individuals from sympatric and allopatric localities (and two diploid individuals of uncertain status) was determined (PopFlag = 0) (Pritchard et al. 2000). Some level of misclassification was allowed with the MIGRPRIOR set at 0.01.
Results
Genotyping: general overview
All analyses were performed with 12 markers in total, nine of which were discriminating for the polyploids. The remaining three markers never presented more than two alleles in any of the inferred polyploids. Although surprising, this is compatible with the alleles present in the putative parental populations for these three loci (Table S4). Low levels of uncertainty were found (<1% disagreement between repeats) and uncertain genotypes were eliminated from further analyses. Polyploid individuals were found only in sympatric localities and classified as hybrids between parthenogens and sexuals (hereafter ‘PS hybrids’). Thirteen individuals presented three alleles only for one marker (Table S1). These individuals may have been triploids with unusually low heterozygosity, or there may have been genotyping errors but it is likely they were aneuploid, since aneuploidy has been observed previously in this genus (Kupriyanova 2009). Given the purpose of this study, we followed a conservative approach and kept these individuals in downstream analyses as diploids, with the specific markers assigned as missing data.
Ploidy determination and the exploratory DAPC identified some putative misidentifications (details in SI). These individuals were kept in the analysis and reclassified according to their genetic determination, followed by revised DAPC analyses (Fig. S3).
Sexual and parthenogenetic species structure
The optimal number of clusters was selected after comparison of the BIC values from DAPC with the best K values in STRUCTURE. When setting clusters for the sexual populations only, K = 7 resulted in the most consistent groups in both the DAPC and STRUCTURE analyses. These clusters corresponded to the recognised species, but also distinguished groups within species. In D. valentini and D. raddei this was probably due to spatial structure, reflected in significant isolation by distance (Figs. S4 and S5). Darevskia mixta consistently fell into two well differentiated and geographically separated groups (D. mixta-1 and D. mixta-2), with an FST distance (FST = 0.25, Table 1) similar to, or even higher than, other recognised species pairs. These subgroups within species were kept separate since they may be relevant to determining the parentage of parthenogens. All sexual species showed heterozygote deficiencies (Fig. 2), most likely due to merging of geographically variable populations.

Expected and observed heterozygosity for all diploid species. Codes are as follows: rud—D. rudis, mix2—D. mixta-2, mix—D. mixta, val—D. valentini population 1, val2—D. valentini population 2, rad—D. raddei population 1, rad2—D. raddei population 2, arm—D. armeniaca, k_arm—D. armeniaca from Kuchak, uni—D. unisexualis, uzz—D. uzzelli. Parthenogens (arm, uni and uzz) are identified with the starred Venus symbol. Colour codes are the same as in Fig. 1
When including the three parthenogenetic species, they were consistently divided into four clusters in DAPC (two clusters for D. armeniaca). Therefore, a K value of 11 was used in the DAPC of the total diploid dataset (sexual + parthenogenetic, including diploids of the sympatric localities). Parthenogens (D. unisexualis, D. uzzelli and D. armeniaca) had higher observed than expected heterozygosity, as predicted due to their hybrid origin (Fig. 2). All parthenogenetic species had private alleles, absent from the sexual taxa and sometimes in high frequencies (e.g. allele 258 represents 53% of the diversity of the C113 marker in D. uzzelli; Table S5).
Asexual parentage inference
STRUCTURE analysis following training on the sexual species (K = 7) clearly showed a mixed ancestry for each of the parthenogenetic genomes (Fig. 3, Table S6). Darevskia armeniaca shared approximately half of its genome with D. mixta-1 and the other half was divided between D. valentini and D. rudis (predominantly from one cluster within each of these sexual species). Similarly, D. uzzelli shared half of its genome with D. raddei and the other half with a mix of D. valentini and D. rudis clusters. The D. unisexualis genome was also shared half with D. raddei and the other half with the D. valentini and D. rudis clusters, in a similar but not identical composition to D. uzzelli. Mitochondrial DNA sequences confirmed previous maternal parent assignments (Fig. S6; see SI for methods). Across all three parthenogens, the contribution from the putative maternal species was closer to 50% (0.36–0.58) than the contribution from the proposed paternal species (0.10–0.45). This is reflected in lower FST between each parthenogen and its maternal parent than its putative paternal ancestor (Table 1).

Bayesian clustering results from STRUCTURE for the sexual (D. raddei, D. mixta-1, D. mixta-2, D. valentini and D. rudis) and asexual (D. armeniaca, D. unisexualis and D. uzzelli) species for K = 7 and with population prior set. D. raddei and D. valentini were each divided into two populations as in Table 1
Origin of parthenogenetic hybrids: number of hybridisation events
Analyses of the distributions of Bruvo distances were performed to assess the number of origins of each parthenogenetic species. A parthenogenetic species with a single origin is expected to display a unimodal distribution whose variance increases with lineage age and might become fragmented due to spatial structure and extinction of lineages. On the other hand, more than one origin might generate a multimodal distribution dependent on sampling of genotypes from the parental populations.
The D. armeniaca distance distribution (Fig. 4, inset A) not only extended over a wider range than the remaining parthenogens, but also was bimodal (with the first peak more marked than the second; Hartigans’ dip test D = 0.0657, p-value < 2.2e−16). In the DAPC analysis (Fig. S7), Kuchak individuals and those from the remaining distribution were separated into two groups, a separation possibly accentuated by the overrepresentation of Kuchak individuals in the dataset. Nevertheless, the presence of a higher number of private alleles (Table S5) in the population from Kuchak, in comparison to the remaining distribution, suggests that this population has had more time to accumulate new alleles than the rest of the distribution. These observations are consistent with D. armeniaca being older than the other parthenogens and possibly having more than one origin for extant clones.

Bruvo pairwise distances among individuals of each of the parthenogenetic species (top panel) and among individuals of each of the polyploid backcrosses types (bottom panel). Facet A corresponds to the pairwise distances within D. armeniaca excluding individuals from Kuchak
Bruvo distances for D. uzzelli were distributed over a range of distances similar to D. armeniaca, there was a significant departure from unimodality (Hartigans’ dip test D = 0.0528, p-value < 2.2e−16) and a significant bimodality. Despite the wide interval of distances, D. uzzelli was the parthenogen with the lowest number of private alleles.
Finally, D. unisexualis had one peak in the Bruvo distance distribution, and the narrowest range of distances among the three parthenogens analysed. Hartigans’ dip test indicated departure from unimodality (D = 0.03065, p-value = 0.002), but the distribution was unimodal at low resolution and ragged at higher resolution; it also indicated multimodality with at least four peaks. A bimodal pattern was recovered in the DAPC analysis. Darevskia unisexualis from Kuchak also had private alleles (Table S5) but, in this species, more private alleles were found in the remaining distribution (unlike D. armeniaca).
Sympatric localities
Polyploid individuals were found in three of the sympatric localities, Kuchak, Sotk and Lchashen. Following ploidy assignment in Kuchak, 17% (27/160) were polyploids and interpreted as parthenogenetic × sexual (PS) hybrids: 3% (5/160) D. armeniaca × D. valentini and 14% (22/160) D. unisexualis × D. valentini. Diploid individuals from Kuchak belonged to the parthenogens D. unisexualis and D. armeniaca, and the sexual D. valentini. One tetraploid was found in Kuchak and another in Sotk, both PS hybrids D. armeniaca × D. valentini (IDs 12176 and 9910, respectively: Table S1). In Sotk, 11 individuals were D. armeniaca × D. valentini triploids and five D. valentini sexual diploids.
In the PCoA analysis of Kuchak and Sotk (Fig. 5a), a total of 277 individuals were included: 160 from Kuchak and 25 from Sotk, the remainder from the general distribution range (Table S1). Darevskia valentini formed a separate cluster from each of the two parthenogens, D. armeniaca and D. unisexualis, and the three clusters were approximately equidistant. Diploid individuals from Kuchak and Sotk mostly overlapped with the distribution of genotypes from elsewhere in the corresponding species’ ranges, with some exceptions (specifically among D. armeniaca and D. valentini from Kuchak). As expected, genotypic variation was greater in the sexual species than the parthenogens.

Principal Coordinate Analysis (PCoA) of the Bruvo distances between individuals from the sympatric localities, including polyploid hybrids. Sympatric localities Sotk and Kuchak are in a, and Lchap and Lchaschen in b. Diploid individuals from non-sympatric localities are represented by the convex hulls, one per species (a D. armeniaca, D. valentini and D. unisexualis; b D. armeniaca, D. unisexualis, D. valentini and D. raddei). Individuals from the sympatric localities are identified as triangles (triploid backcrosses), filled circles (sexual and parthenogenetic diploids) or squares (tetraploids), and in different colour tones whenever from different localities
Triploid D. armeniaca × D. valentini PS hybrids from Kuchak and Sotk fell between their proposed maternal (the parthenogen D. armeniaca) and paternal species (the sexual D. valentini). As expected, these clusters were closer to the parthenogenetic parent, which contributed two out of three alleles. Triploid D. unisexualis × D. valentini PS hybrids from Kuchak behaved similarly, clustering between their proposed maternal (the parthenogen D. unisexualis) and paternal species (the sexual D. valentini). Both tetraploid individuals fell among the remaining PS hybrids of the same cross (Fig. 5a).
Bruvo distances among the PS hybrids were distributed over a narrower interval for D. unisexualis × D. valentini than for D. armeniaca × D. valentini hybrids, even though there were many more D. unisexualis × D. valentini comparisons (Fig. 4, bottom panels). Average pairwise distances were generally larger for the polyploids than for the diploid parthenogens (Fig. 4, top panel), and a pairwise distance of zero was never found among the polyploids.
If the triploid PS hybrids originate via recurrent backcrossing each generation between the asexual females and the sexual males, they are expected to carry only alleles that are shared with their parthenogenetic maternal species (D. armeniaca or D. unisexualis, which are also of hybrid origin) or with the paternal species (D. valentini). As predicted, we found that PS hybrids carried one allele unique to D. valentini and the other (one or) two either unique to the parthenogen (D. unisexualis or D. armeniaca) or shared by the parthenogen and D. valentini. The allelic combination for each marker was normally such that the source of each allele could be easily determined. Only two D. unisexualis × D. valentini triploid PS hybrids (IDs 12182 and 12574) had unique alleles (Table S5), one allele each that was absent from all other species in this study.
In the PCoA with the Lchap and Lchashen (Fig. 5, panel b), only 19 individuals in total were available (Table S1). All D. raddei individuals from these localities clustered with D. raddei from elsewhere in its range, and separately from the D. unisexualis cluster found in Lchap. D. valentini individuals fell close to, but not within the cluster formed by D. valentini individuals from elsewhere, and unexpectedly not very close together. The PS hybrid D. armeniaca × D. valentini found in Lchashen (red triangle on the Fig. 5, panel b) fell within the cluster formed by the PS hybrids D. armeniaca × D. valentini from Sotk when analysed together (results not shown).
Diploids from the sympatric localities were included in the structure analysis and the genomic contributions of parents to these individuals were concordant with the same species from other non-sympatric localities. The proportional contributions from the putative parents were slightly different between D. armeniaca from Kuchak and from elsewhere (Table S6), consistent with weak bimodality in the Bruvo pairwise genetic distance distribution (Fig. 4, top panel) and DAPC. The contributions varied little among Kuchak individuals, indicating the presence of one abundant clone (and generating the peak at low Bruvo distance).
Discussion
Our study focused on Darevskia rock lizards as a model for the origin and dynamics of parthenogenesis in vertebrates. Our results support previous parentage assignments for the parthenogens. Only two lineages act as parents, one consistently as the maternal parent. This pattern is more compatible with the PCH than with the core BH. We present evidence for multiple origins of each parthenogenetic species. Our results also show ongoing hybridisation between parthenogenetic females and sexual males, but no gene flow was detected among the interbreeding species.
The sexual (and diploid) Darevskia lizards analysed here were divided into genetically distinct groups, concordant with the initial assignment based on phenotype. Darevskia mixta individuals were consistently divided into two genetic groups, one with samples from Georgia (D. mixta-1) and the other with samples from Turkey (D. mixta-2). Pairwise FST between these groups was 0.25, similar to the distance between other sexual species pairs, suggesting species-level differentiation.
With structure analysis, we reassessed the parentage inferences made by Darevsky (1967), Fu et al. (2000a, 2000b) and Murphy (2000) and confirmed the hybrid genetic profile of all diploid parthenogenetic species studied. This analysis was performed with the allelic information from 12 microsatellite loci only, so it is likely to have some uncertainty. However, lack of LD among the loci suggests that they are not closely linked and so provide a meaningful estimate of the proportional contributions of parental genomes.
We found that the maternal contribution (confirmed by mtDNA analysis, Fig. S6) for each parthenogen (either D. raddei or D. mixta-1) accounted for ~50% of the nuclear genome of the parthenogenetic species, as expected. On the other hand, the paternal contribution was less clear (Fig. 3). For each of the three parthenogens, there appeared to be a mix of contributions from the sexual species D. valentini and D. rudis. Darevskia valentini was the previously proposed paternal species in all these cases (Darevsky 1967). On the basis of five microsatellite markers, Tarkhnishvili et al. (2017) claimed to provide evidence for the origin of some Darevskia parthenogenetic lineages (e.g. D. armeniaca) from backcross hybridisation between one parthenogenetic female and a male of a sexual species (Tarkhnishvili et al. 2017). However, that would not explain the 50% maternal genomic ancestry found in all parthenogenetic individuals analysed here, since it would imply triploidy and a contribution of ~33%. More parsimonious explanations are available: a prior hybridisation between two sexual individuals of different species, in this case D. rudis and D. valentini (for which there is some evidence; Tarkhnishvili et al. 2013), could have generated a sexual hybrid with a mixed genome that then hybridised with a female of a different sexual species (either D. raddei or D. mixta-1), leading to the origin of an asexual hybrid lineage with contributions from three parental taxa. For this to have happened, the three different species would have to have been in sympatry, a scenario facilitated by the frequent secondary contacts Darevskia taxa experienced during the Pleistocene ice ages (Freitas et al. 2016). Moreover, widespread evidence for gene flow among sexual taxa (Freitas et al. unpublished), including among species of the rudis clade (D. valentini, D. rudis and D. portschinskii) (Tarkhnishvili et al. 2013), further supports this suggestion. Ancient hybridisation appears to fuel speciation events in other taxa as well (e.g. Meier et al. 2017). We cannot rule out contributions from unsampled populations, but we conclude that prior hybridisation is the most likely explanation for our findings.
(1) Is the origin of hybrid asexuality in Darevskia more consistent with the Balance Hypothesis or with the Phylogenetic Constraint Hypothesis?
Under the BH (Moritz et al. 1989), parental species pairwise distances, at the inferred time of origin of the parthenogenetic hybrids, should fall within a constrained interval (Kearney et al. 2009). Any species pair that has passed through that interval could have generated parthenogenetic hybrids but it is likely that only a small proportion of them will give rise to lineages that are now extant. In Darevskia, we used FST pairwise distances calculated from the 12 microsatellite markers. Given the small number of markers used and the potential for homoplasy, distances may be inaccurate, particularly under-estimating longer distances. But, given the young age of the parthenogens (Freitas et al. 2016), current parental distances are not much greater than distances when hybridisation gave rise to them. From these data we cannot assess constraints on the divergence interval during which parthenogens can be formed. However, there are sexual species pairs with estimated distances within the same interval as the sexual parentals and with geographical range overlap (e.g. D. mixta-1/D. rudis in Western Caucasus). No parthenogenetic hybrid between these species has been reported. This conclusion also holds for distances based on mtDNA sequences (Table 1).
The 23 sexual species of Darevskia not included in this study produced no known parthenogenetic hybrid species despite extensive survey effort with this group (Murphy 2000). This pattern is unexpected if phylogenetic distance is the sole determinant of the potential for sexual species to give rise to parthenogenetic hybrids. On the other hand, it is the expected pattern under the PCH. More precise distance estimates, based on more sequence data, and for a larger sample of species, will be needed to investigate this pattern further.
We also confirm that the directionality of the hybridisation events was consistent, D. mixta-1 and D. raddei always acting as maternal species. This pattern is not expected under the core BH. While it is not a necessary feature of the PCH, it can arise because the predisposition of particular lineages to generate parthenogenetic hybrids may be specific to either the maternal or paternal role. If such a predisposition is maintained in the parthenogens, it may help to explain their observed ability to produce viable offspring with the sexual males of their paternal species.
Despite the viability of the offspring between parthenogenetic females and sexual males, the resulting hybrids are polyploid and apparently infertile, incapable of generating a stable parthenogenetic lineage. The infertility of the polyploid hybrids could be explained under the extended BH, which proposes that the addition of a second haploid genome from one of the parental species can disrupt the balance between meiotic disruption and incompatibility that allows the diploid hybrids to reproduce parthenogenetically. However, this hypothesis also supports the alternative possibility (Moritz et al. 1989; Fig. 8), that the addition of a third genome can increase the fecundity and viability of the asexual hybrids (allowing a polyploid lineage to be generated). Similarly, change in ploidy could have either effect on the lineage-specific factors underlying parthenogenesis postulated by the PCH. Thus, the infertility of the Darevskia polyploids does not provide evidence to distinguish between the BH and PCH.
The BH remains the most widely accepted explanation for the origin of vertebrate parthenogenesis (Avise 2008). However, in addition to Darevskia, other hybrid parthenogenetic vertebrates question whether the core BH is widely applicable. The hybridisation events that gave rise to the parthenogenetic Leiolepis (butterfly lizard) are directional and constrained to two clades within the genus (Grismer et al. 2014). In Aspidoscelis, the parthenogenetic hybrids always have a member of the Sexlineatus clade as at least one of the parental species (Darevsky et al. 1985; Reeder et al. 2002). Still in this same genus, the parental pairs of the parthenogenetic hybrids are composed both of species within the same phylogenetic clade (Lemniscatus and Sexlineatus groups), and species from different phylogenetic clades (Cozumela and Tesselata groups), and consequently the distances among the parental pairs can be highly variable (Reeder et al. 2002; and see Janko et al. 2018 for a review showing variable distances between parental pairs of asexual hybrids in Teleostei) suggesting little constraint on the divergence interval. Our results and other studies suggest that the distance between parental species is not as critical as the specific characteristics of the sexual ancestors, and in some cases also the directionality of the hybridisation events. Some constraints on the interval of divergence, as proposed under the BH, must apply. However, lineage-specific factors also appear to be critical to explaining the origin of parthenogenetic hybrid lineages.
(2) Did multiple hybridisation events give rise to each parthenogenetic Darevskia species?
The parthenogenetic Darevskia species were initially described based on their morphological traits (Darevsky 1967; Arakelyan et al. 2011). The number of hybridisation events giving rise to Darevskia extant lineages of parthenogenetic species has been inferred to be one (Darevskia rostombekowi, Ryskov et al. 2017) or more than one (D. dahli, Vergun et al. 2014).
Our data show significant departures from unimodal distance distributions among genotypes for all three parthenogens. Given that these species are all young, this is more likely to be a signal of multiple origins than diversification and fragmentation of a single lineage over time. However, more data and demographic modelling will be needed to distinguish these possibilities. The three species analysed here behave differently in other respects. In D. armeniaca, which has a wide geographic range, the pairwise Bruvo distance interval was wide and several private alleles were observed (most of them in Kuchak). Darevskia unisexualis also has a wide range but its distance interval was narrower and private alleles were not localised. Despite its young age (originated ~100 kya), D. unisexualis is distributed over a relatively wide area (Freitas et al 2016). This rapid colonisation, perhaps combined with competitive interactions between the Darevskia parthenogens and their sexual relatives (Tarkhnishvili et al. 2010), can lead to local extinctions or expansions of clones and so spatial heterogeneity in the parthenogenetic species. The multimodality recovered in the Hartigan’s test could be due to this type of clonal structure.
Darevskia uzzelli has a narrow distribution range but the Bruvo distance interval was as wide as in D. armeniaca, despite it having few private alleles. Darevskia armeniaca may have resulted from older hybridisation events than the other two species. However, a single origin of extant clones followed by mutation accumulation in a patchy spatial distribution could also explain the pattern observed. Evidence in other species (such as sexual trait decay, reviewed in van der Kooi and Schwander 2014) suggests that if the lower number of PS hybrids between D. armeniaca and D. valentini in Kuchak is a result of sexual trait decay, the Kuchak population might be older than the Sotk population.
In localities where the sexual parent species are currently in sympatry, as in Lchashen, no evidence of new parthenogenetic lineages (or any parthenogenetic diploid individual) was found. Thus, there is no evidence that new parthenogenetic hybrids are being generated at present, although a low rate of origin cannot be excluded. The origin of parthenogenetic Darevskia might have been associated with secondary contacts promoted by the last glaciations, as suggested on the basis of phylogeographic patterns and ecological niche models (Freitas et al. 2016).
(3) Is there ongoing backcrossing with gene flow between the asexuals and their sexual parentals?
When hybridising with their sexual ancestors, PS hybrids frequently form new polyploid asexual lineages with hybridisation-induced parthenogenesis (e.g. whiptail lizards Aspidoscelis sp.: Cole et al. 2014; Taylor et al. 2015; Cole et al. 2017, butterfly lizards Leiolepis sp.: Grismer et al. 2014; and the salamander Ambystoma sp.: Bi and Bogart 2010). However, it is also possible that triploid hybrids are formed in each generation and never establish polyploid lineages. Our results are more consistent with recurrent backcrossing between sexual and parthenogenetic Darevskia than with the existence of polyploid asexual lineages. Firstly, the pairwise distance distributions of the backcross polyploid hybrids (PS hybrids) match the pairwise distance distributions of simulated genotypes produced by recurrent backcrossing (Fig. S8). Secondly, we did not recover any PS hybrid pair of individuals with the same combination of genotypes for all markers, which we would find if the PS hybrids reproduced clonally. The rare private alleles carried by the PS hybrids are likely due to limited sampling, or possibly genotyping error.
The reproductive organs of PS hybrids from the populations studied here have been found to be undeveloped (Danielyan et al. 2008). However, some of the triploids analysed here did show apparently normal reproductive organs or evidence of having already laid eggs (such as triploid female ID 10034 from Sotk). Triploid hybrid males with apparently normal reproductive systems have already been described in this locality (Darevsky et al. 1985). Thus, the tetraploid individuals we found are likely to be the result of a cross between a triploid and a diploid individual (either sexual D. valentini or parthenogen D. unisexualis and D. armeniaca). Both observations suggest that some triploids could be partially fertile, although offspring viability is unknown.
Traits required for sexual reproduction will be lost over time in asexual lineages (reviewed in van der Kooi and Schwander 2014), with some reported cases of rapid loss (e.g. Lehmann et al. 2011). In Darevskia, the parthenogenetic lineages are young (around 100 kyr old; Freitas et al. 2016) and the generation of triploid PS hybrids shows that they retain the sexual machinery necessary to mate and produce zygotes with a paternal contribution. However, these functions are expected to decay with lineage age (van der Kooi and Schwander 2014). In Kuchak, we found a high ratio of D. unisexualis × D. valentini PS hybrids relative to D. armeniaca × D. valentini, concordant with previous findings (4:1 ratio in Danielyan et al. 2008). The reproductive pressure inferred from the intensity of copulation marks of D. valentini males on each parthenogenetic species present in Kuchak is the same (Carretero et al. 2018). Therefore, one possible explanation for the high proportion of D. armeniaca × D. valentini PS hybrids found in Sotk, and low proportion of this cross found in Kuchak in relation to the other PS hybrid, is that the D. armeniaca lineage present in Kuchak is older and has lost part of the sexual reproduction machinery. This is concordant with the multimodal Bruvo distance distribution for D. armeniaca, suggesting that it had more than one origin through hybridisation events separated in time and/or space.
Evolutionary consequences of hybridisation and polyploidy in Darevskia
Asexuality in vertebrates can be a stage in the speciation continuum, providing an effective barrier to gene flow when other forms of pre- and post-zygotic reproductive isolation are absent (Janko et al. 2018). We identified many polyploid hybrids (17%) in sympatric locations of parthenogenetic species with their paternal sexual ancestors. Nevertheless, sexual and asexual species in the sympatric localities maintain their distinctiveness, suggesting there is an effective barrier to gene flow.
Parthenogenetic Darevskia species may outcompete their sexual ancestors (Tarkhnishvili et al. 2010), or be sole occupants of habitats suitable for their parentals (Freitas et al. 2016). When in contact, hybridisation between parthenogens and sexual males could reduce the number of sexual offspring in each generation. In this way, sexual populations could experience an additional load, especially when in low abundance relative to parthenogens. This demographic vortex might lead sexual species towards local extinction. Together with the two-fold reproductive output advantage of parthenogens compared with sexuals, hybridisation between the two forms could contribute to the out-performance by some parthenogenetic species of their sexual ancestors.
In summary, our study of Darevskia as a model of vertebrate parthenogenesis questions whether the BH, suggested as a general theory for the origin of hybrid asexuality, is sufficient to explain patterns in the origin of hybrid asexuality. Parthenogenesis in vertebrates is rare and generally originates from hybridisation between specific species pairs with highly variable phylogenetic distances but often consistent maternal parentage. This fits the PCH that the parental species must hold some lineage-dependent characteristics that allow them, when hybridising, to generate offspring capable of reproducing asexually. Identifying the constraining factors underlying the origin of parthenogenetic vertebrates gives us a better chance of understanding how these hybrids use the sexual reproduction machinery to reproduce asexually, escaping the limitations that sex might carry.
Data archiving
Microsatellite genotypes: Dryad https://doi.org/10.5061/dryad.nk410t9 mtDNA sequences: GenBank MN210937 – MN211144, MN211145.
Change history
23 September 2019
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Arakelyan M, Danielyan F, Corti C, Sindaco R, Levinton AE (2011) The herpetofauna of Armenia and Nagorno-Karabakh. Society for the Study of Amphibians and Reptiles: San Francisco
Avise JC (2008) Clonality: the genetics, ecology, and evolution of sexual abstinence in vertebrate animals. Oxford University Press, Oxford: New York, NY
Barraclough TG (2010) Evolving entities: towards a unified framework for understanding diversity at the species and higher levels. Philos Trans R Soc B 365:1801–1813
Bartoš O, Röslein J, Kotusz J, Pačes J, Pekárik L, Petrtýl M et al. (2019) The legacy of sexual ancestors in phenotypic variability, gene expression and homoeolog regulation of asexual hybrids and polyploids. Mol Biol Evol. https://doi.org/10.1093/molbev/msz114
Bell G (1982) The masterpiece of nature: the evolution and genetics of sexuality. University of California Press: Berkeley
Beukeboom LW, Vrijenhoek RC (1998) Evolutionary genetics and ecology of sperm-dependent parthenogenesis. J Evol Biol 11:755–782
Bi K, Bogart JP (2010) Time and time again: unisexual salamanders (genus Ambystoma) are the oldest unisexual vertebrates. BMC Evol Biol 10:238
Birky CW, Barraclough TG (2009) Asexual Speciation. In: Schön I, Martens K, Dijk P (eds) Lost sex: the evolutionary biology of parthenogenesis. Springer Netherlands: Dordrecht, p 201–216
Boudjemadi K, Martin O, Simon J-C, Estoup A (1999) Development and cross-species comparison of microsatellite markers in two lizard species, Lacerta vivipara and Podarcis muralis. Mol Ecol Notes 8:513–525
Bruvo R, Michiels NK, D’souza TG, Schulenburg H (2004) A simple method for the calculation of microsatellite genotype distances irrespective of ploidy level. Mol Ecol 13:2101–2106
Bullini L (1994) Origin and evolution of animal hybrid species. Trends Ecol Evol 9:422–426
Carretero MA, García-Muñoz E, Argaña E, Freitas S, Corti C, Arakelyan M et al. (2018) Parthenogenetic females mate if males are around. An analysis of copulation marks in a Darevskia mixed community. J Nat Hist 52:405–413
Choleva L, Janko K, De Gelas K, Bohlen J, Šlechtová V, Rábová M et al. (2012) Synthesis of clonality and polyploidy in vertebrate animals by hybridization between two sexual species: synthesis of clonality and polyploidy in vertebrate animals. Evolution 66:2191–2203
Clark LV, Jasieniuk M (2011) polysat: an R package for polyploid microsatellite analysis. Mol Ecol Resour 11:562–566
Cole CJ, Taylor HL, Baumann DP, Baumann P (2014) Neaves’ whiptail lizard: the first known tetraploid parthenogenetic tetrapod (Reptilia: Squamata: Teiidae). Breviora 539:1–20
Cole CJ, Taylor HL, Neaves WB, Baumann DP, Newton A, Schnittker R et al. (2017) The second known tetraploid species of parthenogenetic tetrapod (Reptilia: Squamata: Teiidae): description, reproduction, comparisons with ancestral taxa, and origins of multiple clones. Bull Mus Comp Zool 161:285–322
Coyne JA, Orr HA, Futuyma DJ (1988) Do we need a new species concept? Syst Zool 37:190–200
Danielyan F, Arakelyan M, Stepanyan I (2008) Hybrids of Darevskia valentini, D. armeniaca and D. unisexualis from a sympatric population in Armenia. Amphib-Reptil 29:487–504
Darevsky IS, Kupriyanova LA, Uzzell T (1985) Parthenogenesis in reptiles. In: Gans C, Billett F (eds) Biology of the reptilia. Wiley: New York, NY, vol. 15, 411–526
Darevsky I (1967) Rock lizards of the Caucasus: systematics, ecology and phylogenesis of the polymorphic groups of Caucasian rock lizards of the subgenus Archaeolacerta. Nauka, Leningrad, Doctoral Thesis
Darevsky IS, Danielyan F (1968) Diploid and triploid progeny arising from natural mating of parthenogenetic Lacerta armeniaca and L. unisexualis with Bisexual L. saxicola valentini. J Herpetol 2:65
Ernst A (1918) Bastardierung als Ursache der Apogamie im Pflanzenreich. Eine Hypothese zur experimentellen Vererbungs- und Abstammungslehre. Nabu Press: Rudolstadt
Freitas S, Rocha S, Campos J, Ahmadzadeh F, Corti C, Sillero N et al. (2016) Parthenogenesis through the ice ages: a biogeographic analysis of Caucasian rock lizards (genus Darevskia). Mol Phylogenet Evol 102:117–127
Fu J, MacCulloch RD, Murphy RW, Darevsky IS, Tuniyev BS (2000a) Allozyme variation patterns and multiple hybridization origins: clonal variation among four sibling parthenogenetic Caucasian rock lizards. Genetica 108:107–112
Fu J, Murphy RW, Darevsky IS (1997) Toward the phylogeny of Caucasian rock lizards: implications from mitochondrial DNA gene sequences (Reptilia: Lacertidae). Zool J Linn Soc 120:463–477
Fu J, Murphy RW, Darevsky IS, McEachran JD (2000b) Divergence of the cytochrome b gene in the Lacerta raddei complex and its parthenogenetic daughter species: evidence for recent multiple origins. Copeia 2:432–440
Goudet J (1995) FSTAT (Version 1.2): a computer program to calculate F-Statistics. J Hered 86:485–486
Grismer JL, Bauer AM, Grismer LL, Thirakhupt K, Aowphol A, Oaks JR et al. (2014) Multiple origins of parthenogenesis, and a revised species phylogeny for the Southeast Asian butterfly lizards, Leiolepis. Biol J Linn Soc 113:1080–1093
Haigh J (1978) The accumulation of deleterious genes in a population—Muller’s Ratchet. Theor Popul Biol 14:251–267
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9:1322–1332
Janko K (2014) Let us not be unfair to asexuals: their ephemerality may be explained by neutral models without invoking any evolutionary constraints of asexuality. Evolution 68:569–576
Janko K, Pačes J, Wilkinson-Herbots H, Costa RJ, Röslein J, Drozd P et al. (2018) Hybrid asexuality as a primary postzygotic barrier between nascent species: on the interconnection between asexuality, hybridization and speciation. Mol Ecol 1:248–263
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94
Kearney M, Fujita MK, Ridenour J (2009) Lost sex in the reptiles: constraints and correlations. In: Schön I, Martens K, Dijk P (eds) Lost sex. Springer: Netherlands, pp 447–474
Korchagin VI, Badaeva TN, Tokarskaya ON, Martirosyan IA, Darevsky IS, Ryskov AP (2007) Molecular characterization of allelic variants of (GATA)n microsatellite loci in parthenogenetic lizards Darevskia unisexualis (Lacertidae). Gene 392:126–133
Kupriyanova L (2009) Cytogenetic and genetic trends in the evolution of unisexual lizards. Cytogenet Genome Res 127:273–279
Lehmann GUC, Siozios S, Bourtzis K, Reinhold K, Lehmann AW (2011) Thelytokous parthenogenesis and the heterogeneous decay of mating behaviours in a bushcricket (Orthopterida). J Zool Syst Evol Res 49:102–109
Lively CM (2010) A review of red queen models for the persistence of obligate sexual reproduction. J Hered 101:S13–S20
Luijckx P, Ho EKH, Gasim M, Chen S, Stanic A, Yanchus C et al. (2017) Higher rates of sex evolve during adaptation to more complex environments. Proc Natl Acad Sci USA 114:534–539
Lutes AA, Baumann DP, Neaves WB, Baumann P (2011) Laboratory synthesis of an independently reproducing vertebrate species. Proc Natl Acad Sci USA 108:9910–9915
Maynard Smith J (1978) The evolution of sex. Cambridge University Press: New York, NY
McDonald MJ, Rice DP, Desai MM (2016) Sex speeds adaptation by altering the dynamics of molecular evolution. Nature 531:233–236
Meier J, Marques D, Mwaiko S, Wagner CE, Excoffier L, Seehausen O (2017) Ancient hybridization fuels rapid cichlid fish adaptive radiations. Nat Commun. 8:14363
Moritz C, Brown WM, Densmore LD, Vyas S, Donnellan S, Adams M et al. (1989) Genetic diversity and the dynamics of hybrid parthenogenesis in Cnemidophorus (Teiidae) and Heteronotia (Gekkonidae). In: Dawley RM, Bogart JP (eds) Evolution and ecology of unisexual vertebrates. New York State Museum: Albany
Moritz C, Donnellan S, Adams M, Baverstock PR (1989) The origin and evolution of parthenogenesis in Heteronotia binoei (Gekkonidae): extensive genotypic diversity among parthenogens. Evolution 43:994–1003
Moritz C, Uzzell T, Spolsky C, Hotz H, Darevsky I, Kupriyanova L et al. (1992) The maternal ancestry and approximate age of parthenogenetic species of Caucasian rock lizards (Lacerta: Lacertidae). Genetica 87:53–62
Murgia C, Pritchard JK, Kim SY, Fassati A, Weiss RA (2006) Clonal origin and evolution of a transmissible. Cancer Cell 126:477–487
Murphy R (2000) A fine line between sex and unisexuality: the phylogenetic constraints on parthenogenesis in lacertid lizards. Zool J Linn Soc 130:527–549
Murphy RW, Darevsky IS, MacCulloch RD, Fu J, Kupriyanova LA (1996) Evolution of the bisexual species of Caucasian rock lizards: a phylogenetic evaluation of allozyme data. Russ J Herpetol 3:18–31
Otto SP (2009) The evolutionary enigma of sex. Am Nat 174:S1–S14
Pinho C, Sequeira F, Godinho R, Harris DJ, Ferrand N (2004) Isolation and characterization of nine microsatellite loci in Podarcis bocagei (Squamata: Lacertidae). Mol Ecol Notes 4:286–288
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Reeder TW, Cole CJ, Dessauer HC (2002) Phylogenetic relationships of whiptail lizards of the genus Cnemidophorus (Squamata: Teiidae): a test of monophyly, reevaluation of karyotypic evolution, and review of hybrid origins. Am Mus Novit 3365:1–61
Remón N, Vila M, Galán P, Naveira H (2008) Isolation and characterization of polymorphic microsatellite markers in Iberolacerta monticola, and cross-species amplification in Iberolacerta galani and Zootoca vivipara. Mol Ecol Resour 8:1351–1353
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Ryskov AP, Osipov FA, Omelchenko AV, Semyenova SK, Girnyk AE, Korchagin VI et al. (2017) The origin of multiple clones in the parthenogenetic lizard species Darevskia rostombekowi. PLoS ONE12:e0185161
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press
Schwander T, Crespi BJ (2009) Multiple direct transitions from sexual reproduction to apomictic parthenogenesis in Timema stick insects. Evol Int J Org Evol 63:84–103
Sinclair EA, Pramuk JB, Bezy RL, Crandall KA, Sites JW Jr (2010) DNA evidence for nonhybrid origins of parthenogenesis in natural populations of vertebrates. Evolution 64:1346–1357
Sillero N, Argaña E, Freitas S, García-Muñoz E, Arakelian M, Corti C et al. (2018) Short term spatial structure of a lizard (Darevskia sp.) community in Armenia. Acta Herpetol 13:155–163
Shine R, Phillips B, Langkilde T, Lutterschmidt DI, Waye H, Mason RT (2004) Mechanisms and consequences of sexual conflict in garter snakes (Thamnophis sirtalis, Colubridae). Behav Ecol 15:654–660
Tarkhnishvili D, Gavashelishvili A, Avaliani A, Murtskhvaladze M, Mumladze L (2010) Unisexual rock lizard might be outcompeting its bisexual progenitors in the Caucasus. Biol J Linn Soc 101:447–460
Tarkhnishvili D, Murtskhvaladze M, Anderson CL (2017) Coincidence of genotypes at two loci in two parthenogenetic rock lizards: how backcrosses might trigger adaptive speciation. Biol J Linn Soc 121:365–378
Tarkhnishvili D, Murtskhvaladze M, Gavashelishvili A (2013) Speciation in Caucasian lizards: climatic dissimilarity of the habitats is more important than isolation time. Biol J Linn Soc 109:876–892
Taylor HL, Walker JM, Cole CJ, Dessauer HC (2015) Morphological divergence and genetic variation in the triploid parthenogenetic teiid lizard, Aspidoscelis neotesselata. J Herpetol 49:491–501
Trifonov VA, Paoletti A, Barucchi VC, Kalinina T, O’Brien PCM, Ferguson-Smith MA et al. (2015) Comparative chromosome painting and NOR distribution suggest a complex hybrid origin of triploid Lepidodactylus lugubris (Gekkonidae). PLoS ONE 10:e0132380
van der Kooi CJ, Schwander T (2014) On the fate of sexual traits under asexuality. Biol Rev 89:805–819
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Vergun AA, Martirosyan IA, Semyenova SK, Omelchenko AV, Petrosyan VG, Lazebny OE et al. (2014) Clonal diversity and clone formation in the parthenogenetic Caucasian rock lizard Darevskia dahli. PLoS ONE 9:e91674
Weismann A (1889) The significance of sexual reproduction in the theory of natural selection. In: Poulton EB, Shipley AE (eds) Essays upon heredity and kindred biological problems. Clarendon Press: Oxford, p 251–332.
Wetherington JD, Kotora KE, Vrijenhoek RC (1987) A test of the spontaneous heterosis hypothesis for unisexual vertebrates. Evolution 41:721–731
Wellenreuther M, Runemark A, Svensson EI, Hansson B (2009) Isolation and characterization of polymorphic microsatellite loci for the Skyros wall lizard Podarcis gaigeae (Squamata: Lacertidae). Mol Ecol Resour 9:1005–1008
Acknowledgements
This work was supported by the project “Preserving Armenian biodiversity: Joint Portuguese – Armenian program for training in modern conservation biology” of Gulbenkian Foundation (Portugal) and the FCT grant SFRH/BD/81483/2011. The authors thank Anna Vardanyan, Claudia Corti and Elena Argaña for field assistance, Andy Krupa, Gavin Horsburgh and Susana Lopes for their assistance with the genotyping, and Steeves Buckland and Óscar Mira for their suggestions in the methods. We also thank Karel Janko and an anonymous reviewer for comments on previous versions of the manuscript. MA was supported by SCS MES RA - RFFR 18 RF-132 project. Capture permits were provided by the Ministry of Nature Protection of Republic Armenia Code 5/22.1/51043 and lizard handling followed the ethical guidelines of Yerevan State University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Freitas, S.N., Harris, D.J., Sillero, N. et al. The role of hybridisation in the origin and evolutionary persistence of vertebrate parthenogens: a case study of Darevskia lizards. Heredity 123, 795–808 (2019). https://doi.org/10.1038/s41437-019-0256-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-019-0256-5
This article is cited by
-
Genotypic similarities among the parthenogenetic Darevskia rock lizards with different hybrid origins
BMC Evolutionary Biology (2020)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}