
Host EPAC1 Modulates Rickettsial Adhesion to Vascular Endothelial Cells via Regulation of ANXA2 Y23 Phosphorylation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
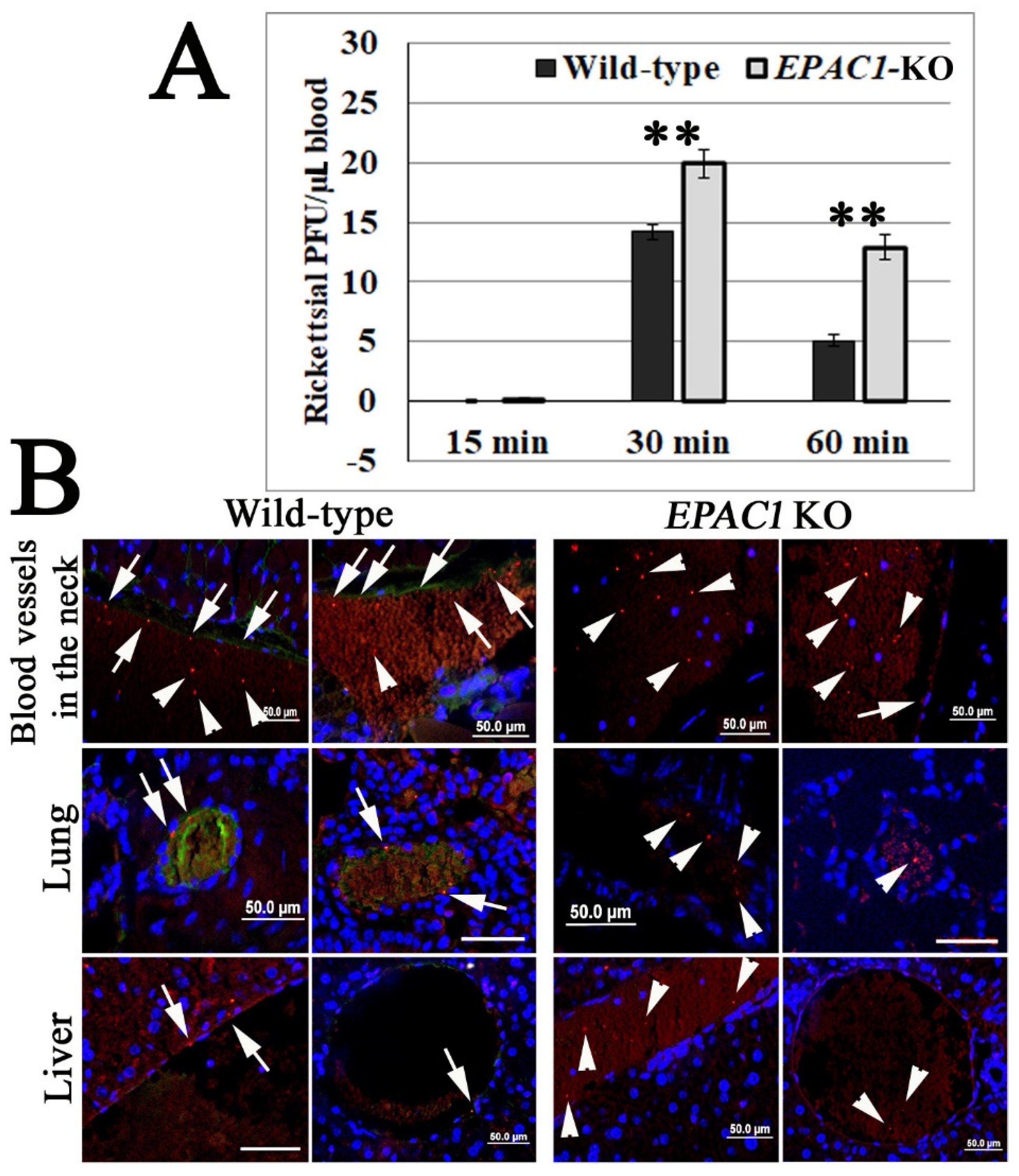
2.1. In Vivo Corroborating Rickettsial Adhesion to EC Surfaces in an EPAC1-Dependent Manner
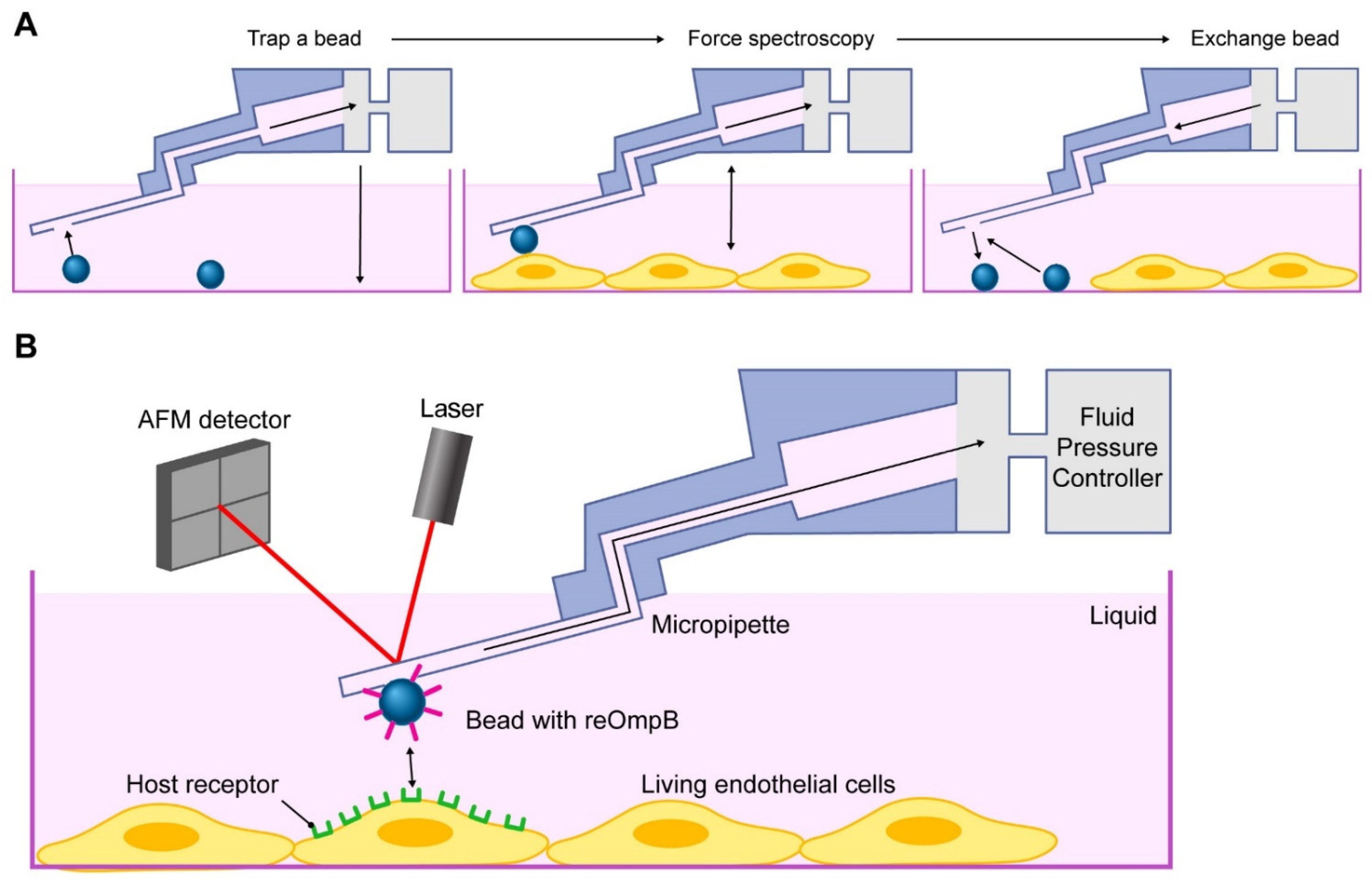
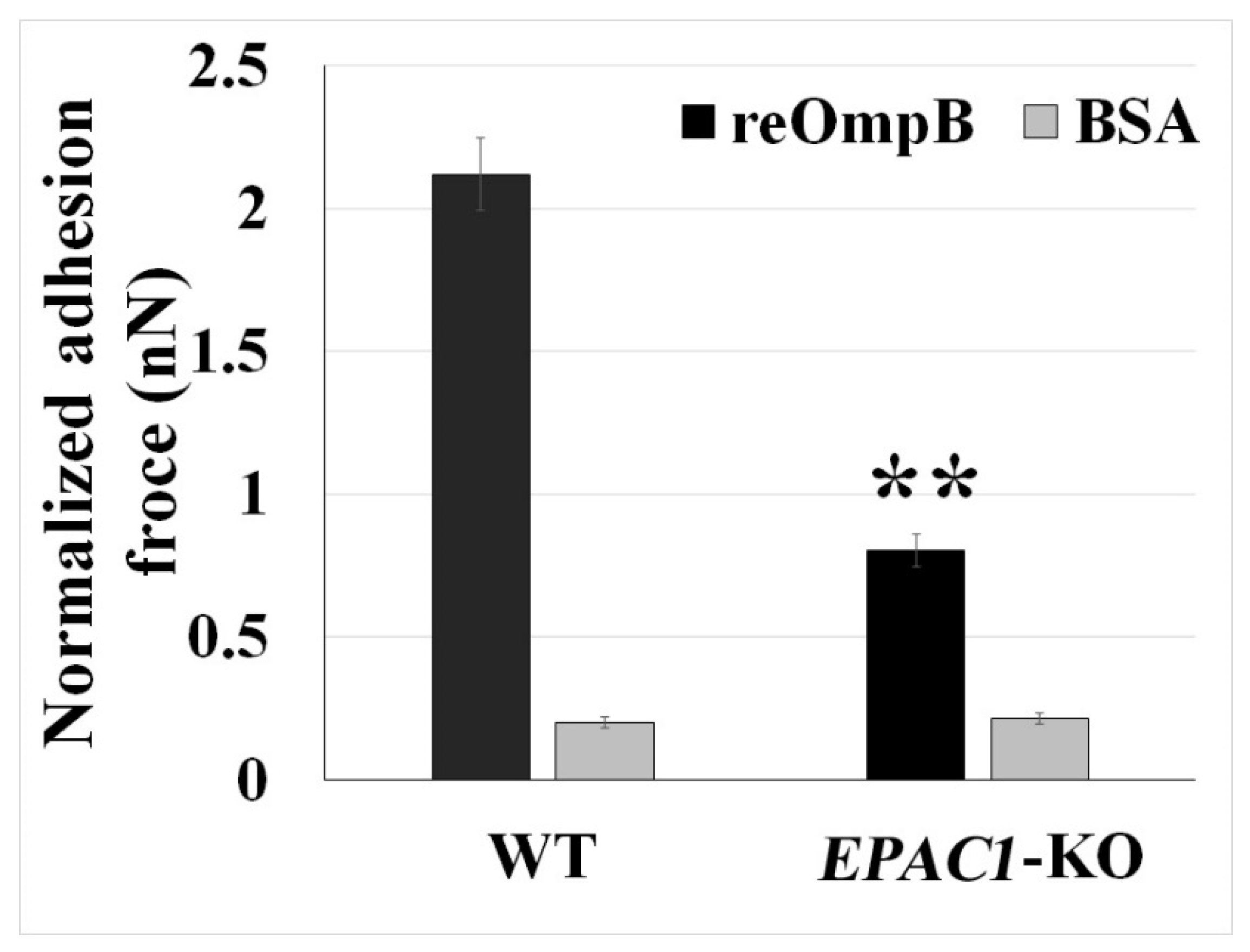
2.2. Rickettsial OmpB Shows a Host EPAC1-Dependent Binding Strength on the Surface of a Living EC
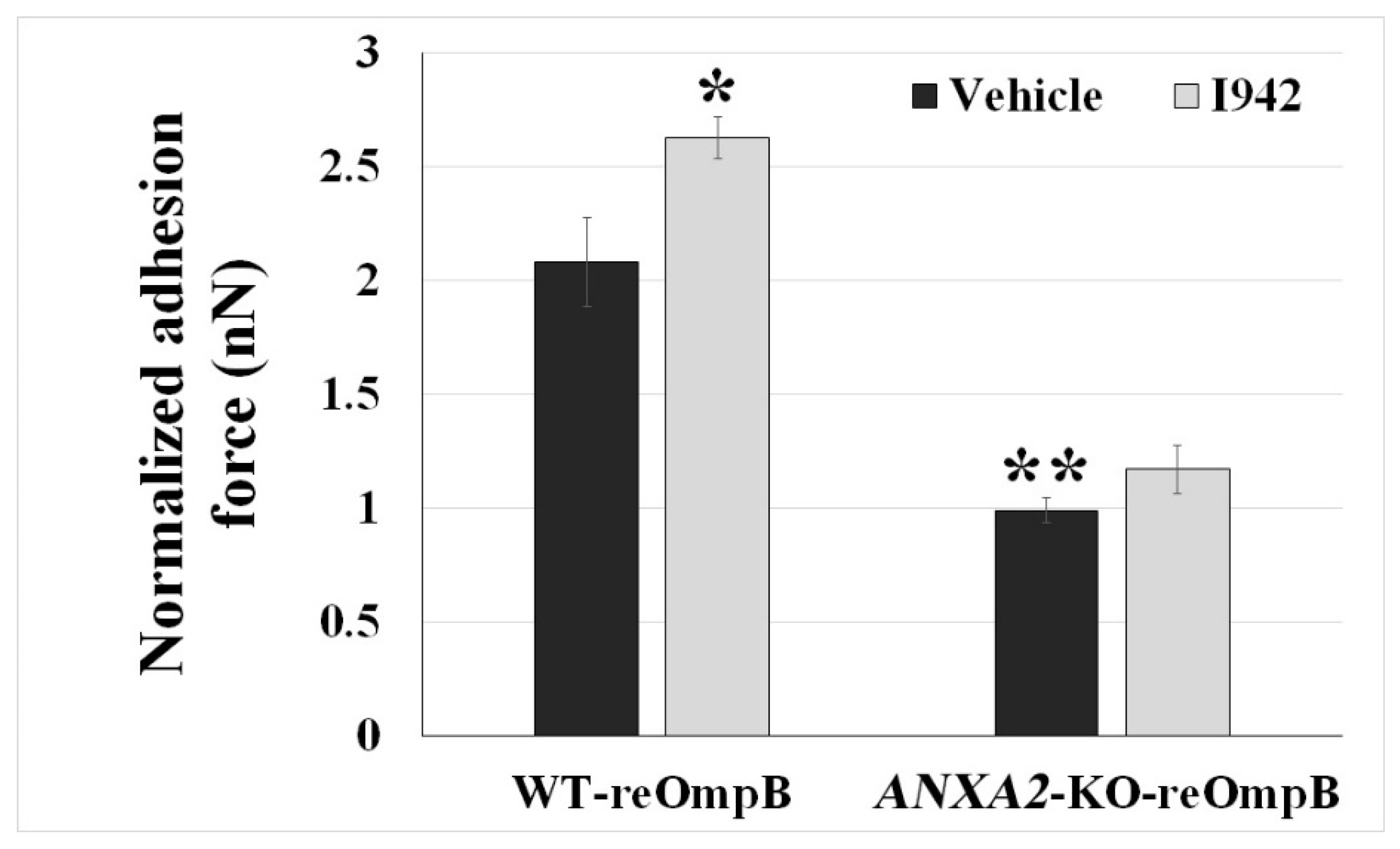
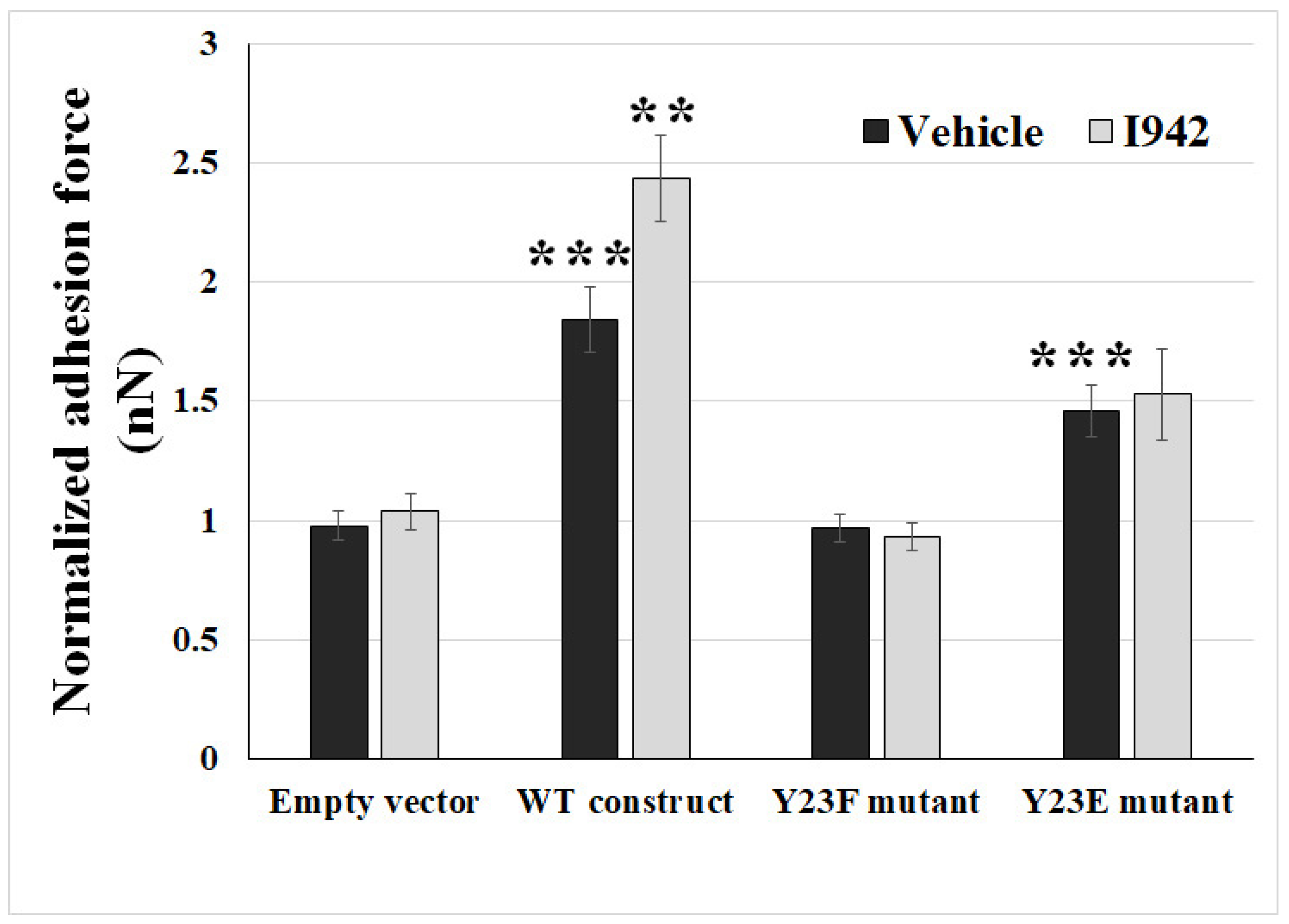
2.3. Ectopic Expression of Phosphodefective and Phosphomimic Mutants Replacing Y23 of ANXA2 in ANXA2-KO BMECs Results in the Display of Different Binding Force to reOmpB in Response to the Activation of EPAC1
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Rickettsia and Cell Culture
4.3. Antibodies and Other Reagents
4.4. Cell Transfection
4.5. WT Construct Sequence (1020 bps)
4.6. Mouse In Vivo, Anatomically-Based, Quantitative Rickettsial Adhesion Measurement System
4.7. FluidFM Adhesion Assay
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dumler, J.S.; Walker, D.H. Rocky Mountain spotted fever-changing ecology and persisting virulence. N. Engl. J. Med. 2005, 353, 551–553. [Google Scholar] [CrossRef]
- Chapman, A.S.; Murphy, S.M.; Demma, L.J.; Holman, R.C.; Curns, A.T.; McQuiston, J.H.; Krebs, J.W.; Swerdlow, D.L. Rocky mountain spotted fever in the United States, 1997–2002. Ann. N. Y. Acad. Sci. 2006, 1078, 154–155. [Google Scholar] [CrossRef]
- Walker, D.H.; Paddock, C.D.; Dumler, J.S. Emerging and re-emerging tick-transmitted rickettsial and ehrlichial infections. Med. Clin. N. Am. 2008, 92, 1345–1361. [Google Scholar] [CrossRef] [PubMed]
- Drexler, N.A.; Dahlgren, F.S.; Heitman, K.N.; Massung, R.F.; Paddock, C.D.; Behravesh, C.B. National Surveillance of Spotted Fever Group Rickettsioses in the United States, 2008–2012. Am. J. Trop. Med. Hyg. 2016, 94, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.B.; Bechelli, J.; Smalley, C.; Karim, S.; Walker, D.H. Vector Tick Transmission Model of Spotted Fever Rickettsiosis. Am. J. Pathol. 2019, 189, 115–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamason, R.L.; Bastounis, E.; Kafai, N.M.; Serrano, R.; Del Álamo, J.C.; Theriot, J.A.; Welch, M.D. Rickettsia Sca4 Reduces Vinculin-Mediated Intercellular Tension to Promote Spread. Cell 2016, 167, 670–683.e10. [Google Scholar] [CrossRef] [Green Version]
- Whitman, T.J.; Richards, A.L.; Paddock, C.D.; Tamminga, C.L.; Sniezek, P.J.; Jiang, J.; Byers, D.K.; Sanders, J.W. Rickettsia parkeri infection after tick bite, Virginia. Emerg. Infect Dis. 2007, 13, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.G.; Riley, S.P.; Martinez, J.J. Adherence to and invasion of host cells by spotted Fever group rickettsia species. Front. Microbiol. 2010, 1, 139. [Google Scholar] [CrossRef] [Green Version]
- Walker, D.H.; Ismail, N. Emerging and re-emerging rickettsioses: Endothelial cell infection and early disease events. Nat. Rev. Genet. 2008, 6, 375–386. [Google Scholar] [CrossRef]
- Feng, H.M.; Wen, J.; Walker, D.H. Rickettsia australis infection: A murine model of a highly invasive vasculopathic rickettsiosis. Am. J. Pathol. 1993, 142, 1471–1482. [Google Scholar]
- Valbuena, G.; Walker, D.H. Infection of the endothelium by members of the order Rickettsiales. Thromb. Haemost. 2009, 102, 1071–1079. [Google Scholar]
- Paris, D.H.; Dumler, J.S. State of the art of diagnosis of rickettsial diseases: The use of blood specimens for diagnosis of scrub typhus, spotted fever group rickettsiosis, and murine typhus. Curr. Opin. Infect. Dis. 2016, 29, 433–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Openshaw, J.J.; Swerdlow, D.L.; Krebs, J.W.; Holman, R.C.; Mandel, E.; Harvey, A.; Haberling, D.; Massung, R.F.; McQuiston, J.H. Rocky mountain spotted fever in the United States, 2000–2007: Interpreting contemporary increases in incidence. Am. J. Trop. Med. Hyg. 2010, 83, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Botelho-Nevers, E.; Socolovschi, C.; Raoult, D.; Parola, P. Treatment of Rickettsia spp. infections: A review. Expert. Rev. Anti. Infect 2012, 10, 1425–1437. [Google Scholar] [CrossRef] [PubMed]
- de Sousa, R.; Nóbrega, S.D.; Bacellar, F.; Torgal, J. Mediterranean spotted fever in Portugal: Risk factors for fatal outcome in 105 hospitalized patients. Ann. N. Y. Acad. Sci. 2003, 990, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Parola, P.; Socolovschi, C.; Jeanjean, L.; Bitam, I.; Fournier, P.E.; Sotto, A.; Labauge, P.; Raoult, D. Warmer weather linked to tick attack and emergence of severe rickettsioses. PLoS Negl. Trop. Dis. 2008, 2, e338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salje, J.; Weitzel, T.; Newton, P.N.; Varghese, G.M.; Day, N. Rickettsial infections: A blind spot in our view of neglected tropical diseases. PLoS Negl. Trop. Dis. 2021, 15, e0009353. [Google Scholar] [CrossRef]
- Kim, H.K.; Premaratna, R.; Missiakas, D.M.; Schneewind, O. O antigen is the target of bactericidal Weil-Felix antibodies. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef] [Green Version]
- Rennoll, S.A.; Rennoll-Bankert, K.E.; Guillotte, M.L.; Lehman, S.S.; Driscoll, T.P.; Beier-Sexton, M.; Rahman, M.S.; Gillespie, J.J.; Azad, A.F. The cat flea (Ctenocephalides felis) immune deficiency signaling pathway regulates rickettsia typhi infection. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef] [Green Version]
- Hillman, R.D.; Baktash, Y.M.; Martinez, J.J. OmpA-mediated rickettsial adherence to and invasion of human endothelial cells is dependent upon interaction with α2β1 integrin. Cell Microbiol. 2013, 15, 727–741. [Google Scholar] [CrossRef] [Green Version]
- Felsheim, R.F.; Kurtti, T.J.; Munderloh, U.G. Genome sequence of the endosymbiont Rickettsia peacockii and comparison with virulent Rickettsia rickettsii: Identification of virulence factors. PLoS ONE 2009, 4, e8361. [Google Scholar] [CrossRef] [PubMed]
- Suwanbongkot, C.; Langohr, I.M.; Harris, E.K.; Dittmar, W.; Christofferson, R.C.; Macaluso, K.R. Spotted fever group. Infect Immun 2019, 87. [Google Scholar] [CrossRef] [Green Version]
- Raker, V.K.; Becker, C.; Steinbrink, K. The cAMP pathway as therapeutic target in autoimmune and inflammatory diseases. Front. Immunol. 2016, 7, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beavo, J.A.; Brunton, L.L. Cyclic nucleotide research—Still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Lefkimmiatis, K.; Zaccolo, M. cAMP signaling in subcellular compartments. Pharmacol. Ther. 2014, 143, 295–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gold, M.G.; Gonen, T.; Scott, J.D. Local cAMP signaling in disease at a glance. J. Cell Sci. 2013, 126, 4537–4543. [Google Scholar] [CrossRef] [Green Version]
- Di Benedetto, G.; Pendin, D.; Greotti, E.; Pizzo, P.; Pozzan, T. Ca2+ and cAMP cross-talk in mitochondria. J. Physiol. 2014, 592, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Hofer, A.M. Interactions between calcium and cAMP signaling. Curr. Med. Chem. 2012, 19, 5768–5773. [Google Scholar] [CrossRef]
- Gong, B.; Shelite, T.; Mei, F.C.; Ha, T.; Hu, Y.; Xu, G.; Chang, Q.; Wakamiya, M.; Ksiazek, T.G.; Boor, P.J.; et al. Exchange protein directly activated by cAMP plays a critical role in bacterial invasion during fatal rickettsioses. Proc. Natl. Acad. Sci. USA 2013, 110, 19615–19620. [Google Scholar] [CrossRef] [Green Version]
- McDonough, K.A.; Rodriguez, A. The myriad roles of cyclic AMP in microbial pathogens: From signal to sword. Nat. Rev. Genet. 2011, 10, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Tao, X.; Mei, F.; Agrawal, A.; Peters, C.J.; Ksiazek, T.G.; Cheng, X.; Tseng, C.T. Blocking of exchange proteins directly activated by cAMP leads to reduced replication of Middle East respiratory syndrome coronavirus. J. Virol. 2014, 88, 3902–3910. [Google Scholar] [CrossRef] [Green Version]
- Xayarath, B.; Freitag, N.E. Uncovering the nonessential nature of an essential second messenger. Cell Host Microbe 2015, 17, 731–732. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Hill, K.K.; Filak, H.; Mogan, J.; Knowles, H.; Zhang, B.; Perraud, A.L.; Cambier, J.C.; Lenz, L.L. MPYS is required for IFN response factor 3 activation and type I IFN production in the response of cultured phagocytes to bacterial second messengers cyclic-di-AMP and cyclic-di-GMP. J. Immunol. 2011, 187, 2595–2601. [Google Scholar] [CrossRef] [Green Version]
- Colonne, P.M.; Winchell, C.G.; Voth, D.E. Hijacking host cell highways: Manipulation of the host actin cytoskeleton by obligate intracellular bacterial pathogens. Front. Cell Infect. Microbiol. 2016, 6, 107. [Google Scholar] [CrossRef] [Green Version]
- Borland, G.; Smith, B.O.; Yarwood, S.J. EPAC proteins transduce diverse cellular actions of cAMP. Br. J. Pharm. 2009, 158, 70–86. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Ji, Z.; Tsalkova, T.; Mei, F. Epac and PKA: A tale of two intracellular cAMP receptors. Acta Biochim. Biophys. Sin. 2008, 40, 651–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef]
- Kooistra, M.R.; Corada, M.; Dejana, E.; Bos, J.L. Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett. 2005, 579, 4966–4972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullere, X.; Shaw, S.K.; Andersson, L.; Hirahashi, J.; Luscinskas, F.W.; Mayadas, T.N. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood 2005, 105, 1950–1955. [Google Scholar] [CrossRef] [Green Version]
- Fukuhara, S.; Sakurai, A.; Sano, H.; Yamagishi, A.; Somekawa, S.; Takakura, N.; Saito, Y.; Kangawa, K.; Mochizuki, N. Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol. Cell Biol. 2005, 25, 136–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajjar, K.A. The biology of annexin A2: From vascular fibrinolysis to innate immunity. Trans. Am. Clin. Clim. Assoc. 2015, 126, 144–155. [Google Scholar]
- Gerke, V.; Moss, S.E. Annexins: From structure to function. Physiol. Rev. 2002, 82, 331–371. [Google Scholar] [CrossRef]
- Stenos, J.; Walker, D.H. The rickettsial outer-membrane protein A and B genes of Rickettsia australis, the most divergent rickettsia of the spotted fever group. Int. J. Syst. Evol. Microbiol. 2000, 50, 1775–1779. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Zhang, W.; Chang, Q.; Su, Z.; Gong, D.; Zhou, Y.; Xiao, J.; Drelich, A.; Liu, Y.; Popov, V. A new role for host annexin A2 in establishing bacterial adhesion to vascular endothelial cells: Lines of evidence from atomic force microscopy and an in vivo study. Lab. Investig. 2019, 99, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Drelich, A.; Yu, S.; Chang, Q.; Gong, D.; Zhou, Y.; Qu, Y.; Yuan, Y.; Su, Z.; Qiu, Y. Exchange protein directly activated by cAMP plays a critical role in regulation of vascular fibrinolysis. Life Sci. 2019, 221, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dorig, P.; Ossola, D.; Truong, A.M.; Graf, M.; Stauffer, F.; Voros, J.; Zambelli, T. Exchangeable colloidal AFM probes for the quantification of irreversible and long-term interactions. Biophys. J. 2013, 105, 463–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, Y.G.-Y.; Riley, S.P.; Chen, E.; Martinez, J.J. Molecular basis of immunity to rickettsial infection conferred through outer membrane protein B. Infect. Immun. 2011, 79, 2303–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, Y.G.; Cardwell, M.M.; Hermanas, T.M.; Uchiyama, T.; Martinez, J.J. Rickettsial outer-membrane protein B (rOmpB) mediates bacterial invasion through Ku70 in an actin, c-Cbl, clathrin and caveolin 2-dependent manner. Cell Microbiol. 2009, 11, 629–644. [Google Scholar] [CrossRef] [Green Version]
- Gomand, F.; Borges, F.; Guerin, J.; El-Kirat-Chatel, S.; Francius, G.; Dumas, D.; Burgain, J.; Gaiani, C. Adhesive interactions between lactic acid bacteria and β-Lactoglobulin: Specificity and impact on bacterial location in whey protein isolate. Front. Microbiol. 2019, 10, 1512. [Google Scholar] [CrossRef] [Green Version]
- Wiejak, J.; van Basten, B.; Luchowska-Stańska, U.; Hamilton, G.; Yarwood, S.J. The novel exchange protein activated by cyclic AMP 1 (EPAC1) agonist, I942, regulates inflammatory gene expression in human umbilical vascular endothelial cells (HUVECs). Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Drelich, A.; Judy, B.; He, X.; Chang, Q.; Yu, S.; Li, X.; Lu, F.; Wakamiya, M.; Popov, V.; Zhou, J. Exchange protein directly activated by cAMP modulates ebola virus uptake into vascular endothelial cells. Viruses 2018, 10, 563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, H.; Mohamed, H.; Boulton, S.; Huang, J.; Wang, P.; Chen, H.; Zhou, J.; Luchowska-Stańska, U.; Jentsch, N.G.; Armstrong, A.L.; et al. Mechanism of Action of an EPAC1-Selective Competitive Partial Agonist. J. Med. Chem. 2020, 63, 4762–4775. [Google Scholar] [CrossRef] [PubMed]
- Rescher, U.; Ludwig, C.; Konietzko, V.; Kharitonenkov, A.; Gerke, V. Tyrosine phosphorylation of annexin A2 regulates Rho-mediated actin rearrangement and cell adhesion. J. Cell Sci. 2008, 121, 2177–2185. [Google Scholar] [CrossRef] [Green Version]
- Uchiyama, T.; Kawano, H.; Kusuhara, Y. The major outer membrane protein rOmpB of spotted fever group rickettsiae functions in the rickettsial adherence to and invasion of Vero cells. Microbes Infect. 2006, 8, 801–809. [Google Scholar] [CrossRef]
- Brandherm, I.; Disse, J.; Zeuschner, D.; Gerke, V. cAMP-induced secretion of endothelial von Willebrand factor is regulated by a phosphorylation/dephosphorylation switch in annexin A2. Blood 2013, 122, 1042–1051. [Google Scholar] [CrossRef] [Green Version]
- He, K.L.; Sui, G.; Xiong, H.; Broekman, M.J.; Huang, B.; Marcus, A.J.; Hajjar, K.A. Feedback regulation of endothelial cell surface plasmin generation by PKC-dependent phosphorylation of annexin A2. J. Biol. Chem. 2011, 286, 15428–15439. [Google Scholar] [CrossRef] [Green Version]
- Valapala, M.; Vishwanatha, J.K. Lipid raft endocytosis and exosomal transport facilitate extracellular trafficking of annexin A2. J. Biol. Chem. 2011, 286, 30911–30925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deora, A.B.; Kreitzer, G.; Jacovina, A.T.; Hajjar, K.A. An annexin 2 phosphorylation switch mediates p11-dependent translocation of annexin 2 to the cell surface. J. Biol. Chem. 2004, 279, 43411–43418. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, D.; Nakayama, Y.; Horimoto, S.; Kuga, T.; Ikeda, K.; Kasahara, K.; Yamaguchi, N. Involvement of Golgi-associated Lyn tyrosine kinase in the translocation of annexin II to the endoplasmic reticulum under oxidative stress. Exp. Cell Res. 2006, 312, 1205–1217. [Google Scholar] [CrossRef]
- Zheng, L.; Foley, K.; Huang, L.; Leubner, A.; Mo, G.; Olino, K.; Edil, B.H.; Mizuma, M.; Sharma, R.; Le, D.T.; et al. Tyrosine 23 phosphorylation-dependent cell-surface localization of annexin A2 is required for invasion and metastases of pancreatic cancer. PLoS ONE 2011, 6, e19390. [Google Scholar] [CrossRef] [Green Version]
- Ling, Q.; Jacovina, A.T.; Deora, A.; Febbraio, M.; Simantov, R.; Silverstein, R.L.; Hempstead, B.; Mark, W.H.; Hajjar, K.A. Annexin II regulates fibrin homeostasis and neoangiogenesis in vivo. J. Clin. Investig. 2004, 113, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhou, C.; Su, Z.; Chang, Q.; Qiu, Y.; Bei, J.; Gaitas, A.; Xiao, J.; Drelich, A.; Khanipov, K.; et al. Endothelial exosome plays functional role during rickettsial infection. mBio 2021. [Google Scholar] [CrossRef] [PubMed]
- Coupling, C. TechNote 205. Available online: https://www.bangslabs.com/sites/default/files/imce/docs/TechNote%20205%20Web.pdf (accessed on 20 August 2021).
- Gounaris, A.D.; Perlmann, G.E. Succinylation of pepsinogen. J. Biol. Chem. 1967, 242, 2739–2745. [Google Scholar] [CrossRef]
- Sader, J.E.; Sanelli, J.A.; Adamson, B.D.; Monty, J.P.; Wei, X.; Crawford, S.A.; Friend, J.R.; Marusic, I.; Mulvaney, P.; Bieske, E.J. Spring constant calibration of atomic force microscope cantilevers of arbitrary shape. Rev. Sci. Instrum. 2012, 83, 103705. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, Z.; Shelite, T.R.; Qiu, Y.; Chang, Q.; Wakamiya, M.; Bei, J.; He, X.; Zhou, C.; Liu, Y.; Nyong, E.; et al. Host EPAC1 Modulates Rickettsial Adhesion to Vascular Endothelial Cells via Regulation of ANXA2 Y23 Phosphorylation. Pathogens 2021, 10, 1307. https://doi.org/10.3390/pathogens10101307
Su Z, Shelite TR, Qiu Y, Chang Q, Wakamiya M, Bei J, He X, Zhou C, Liu Y, Nyong E, et al. Host EPAC1 Modulates Rickettsial Adhesion to Vascular Endothelial Cells via Regulation of ANXA2 Y23 Phosphorylation. Pathogens. 2021; 10(10):1307. https://doi.org/10.3390/pathogens10101307
Chicago/Turabian StyleSu, Zhengchen, Thomas R. Shelite, Yuan Qiu, Qing Chang, Maki Wakamiya, Jiani Bei, Xi He, Changcheng Zhou, Yakun Liu, Emmanuel Nyong, and et al. 2021. "Host EPAC1 Modulates Rickettsial Adhesion to Vascular Endothelial Cells via Regulation of ANXA2 Y23 Phosphorylation" Pathogens 10, no. 10: 1307. https://doi.org/10.3390/pathogens10101307