
A Small Peptide Increases Drug Delivery in Human Melanoma Cells

 , ,
, ,  , , , and
, , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Lines
2.3. Peptide Array Synthesis
2.4. Peptide Array Cell Binding Assay
2.5. FITC Labeled-Peptide Synthesis and Purification
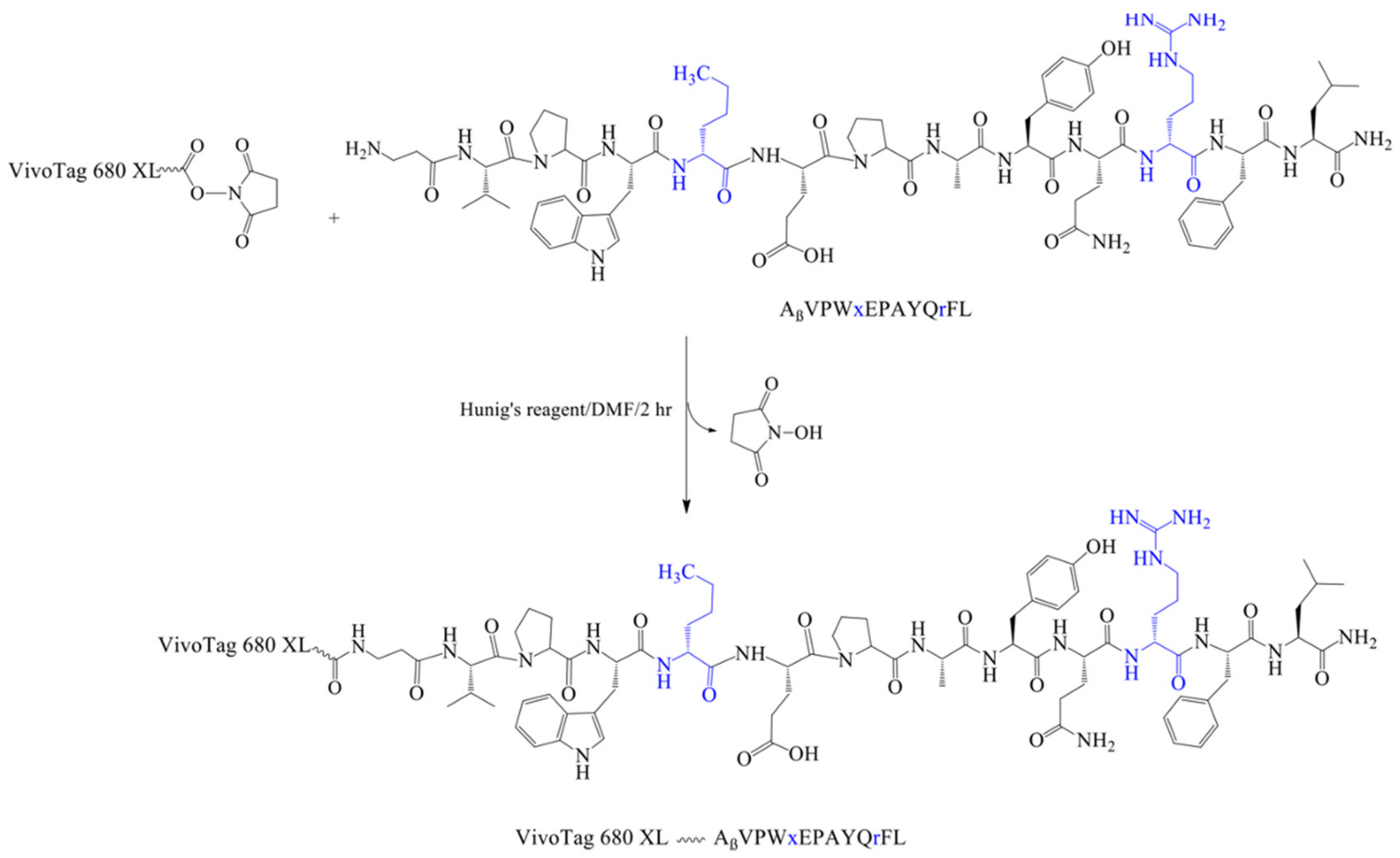
2.6. Synthesis of VivoTag-KK-11 Peptide
2.7. Fluorescence Microscopy and Imaging
2.8. In Vitro Cellular Uptake Analysis
2.9. Serum Stability
2.10. In Vitro Cytotoxicity Detected by MTT Colorimetric Assay
2.11. Ex Vivo and In Vivo Imaging
2.12. In Vivo Xenograft Melanoma Mouse Model
2.13. Statistical Analyses
3. Results
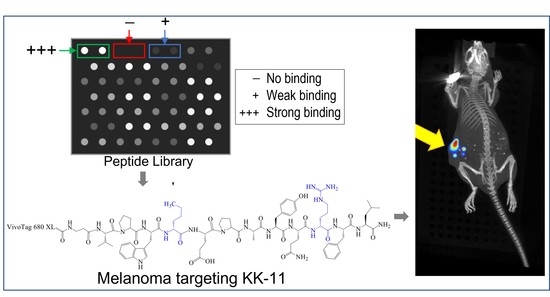
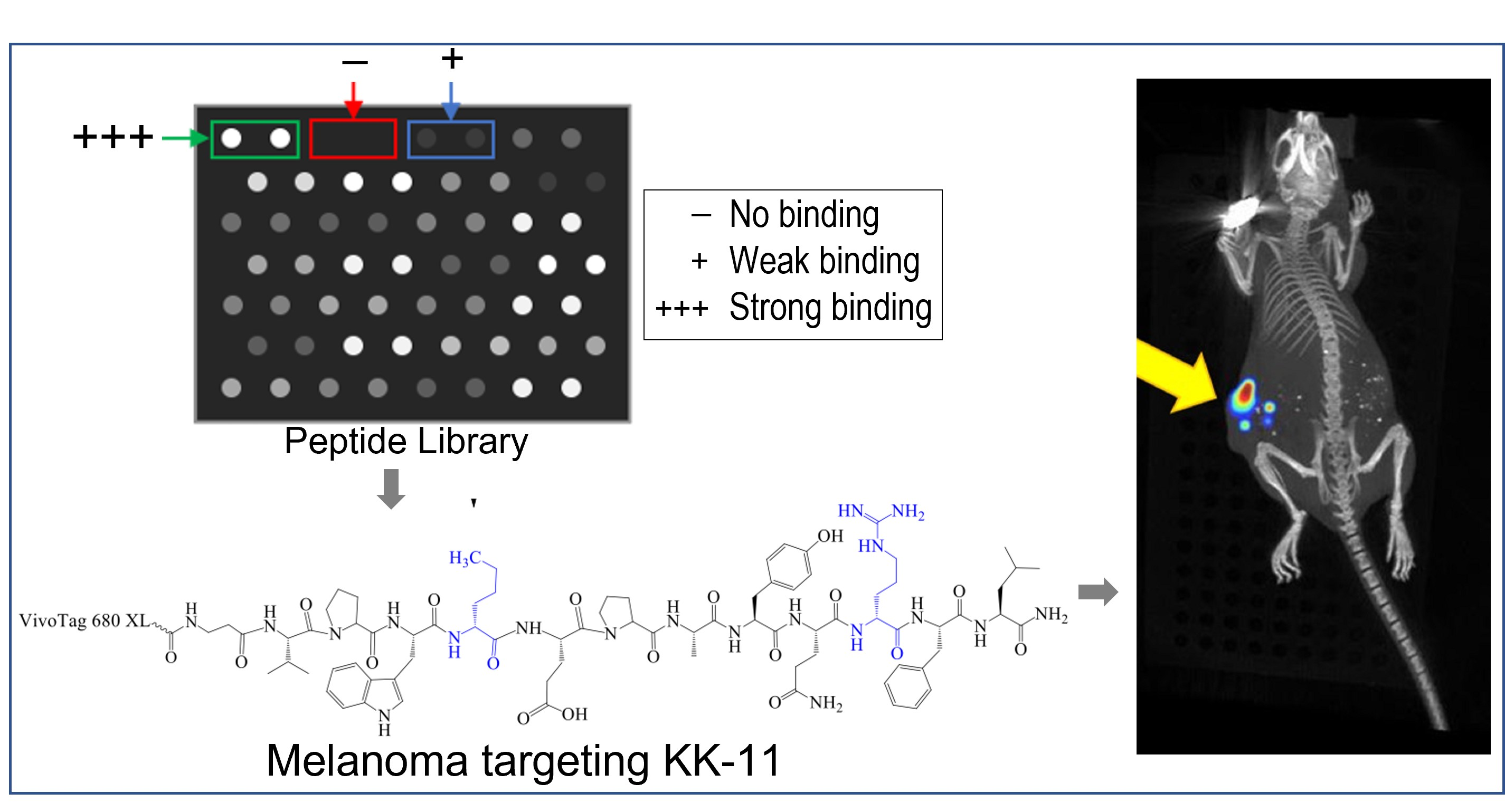
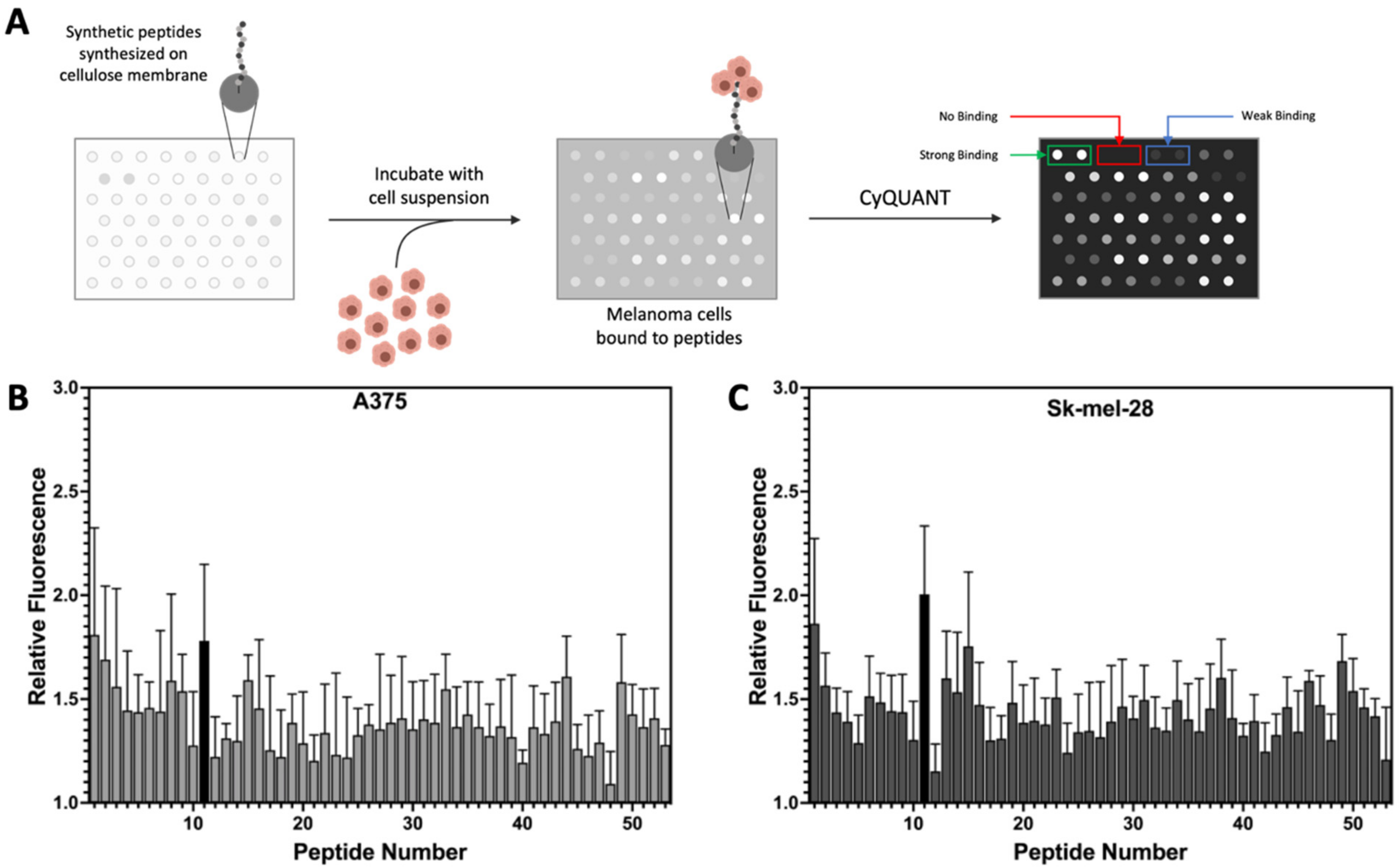
3.1. Peptide Library Synthesis and Screening
3.2. Synthesis of Soluble Fluorescently Labeled Peptides
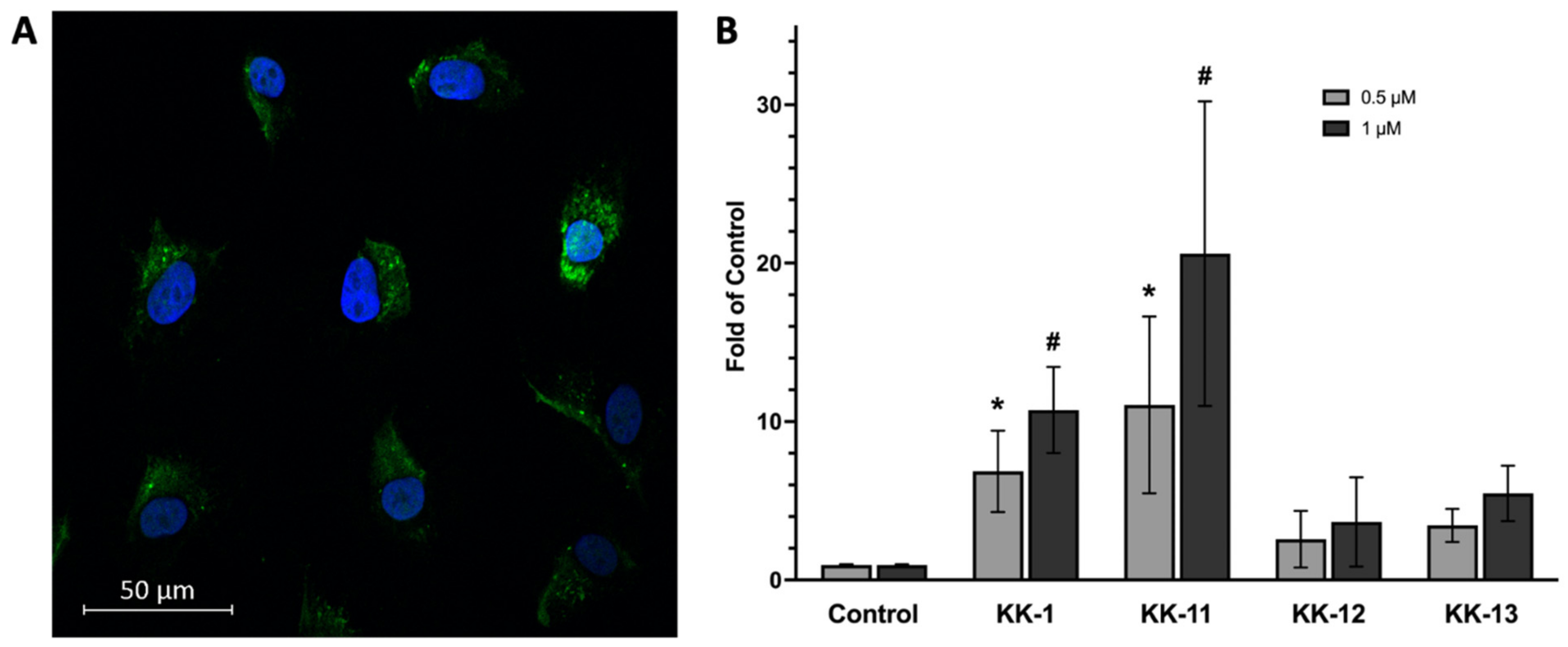
3.3. Uptake of Select Peptides by Melanoma Cells
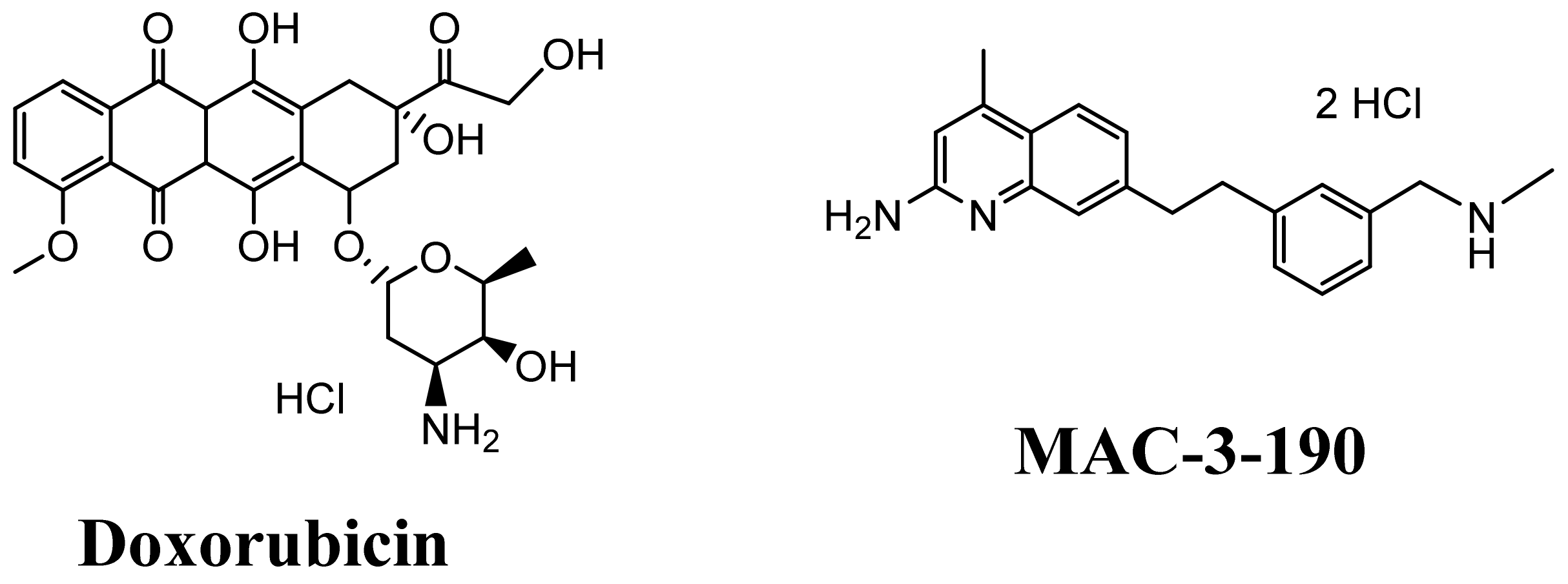
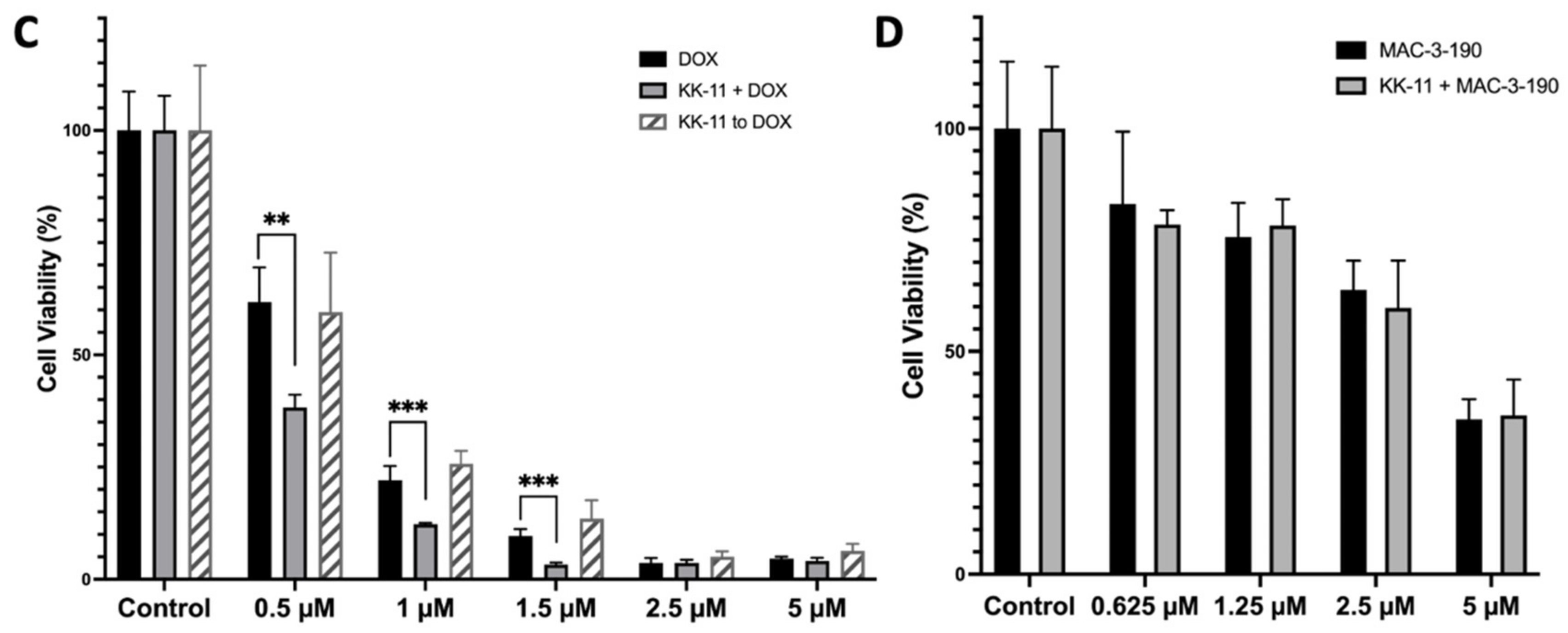
3.4. KK-11 Co-Treatment Significantly Enhanced the Cytotoxicity of DOX in Human Melanoma Cells
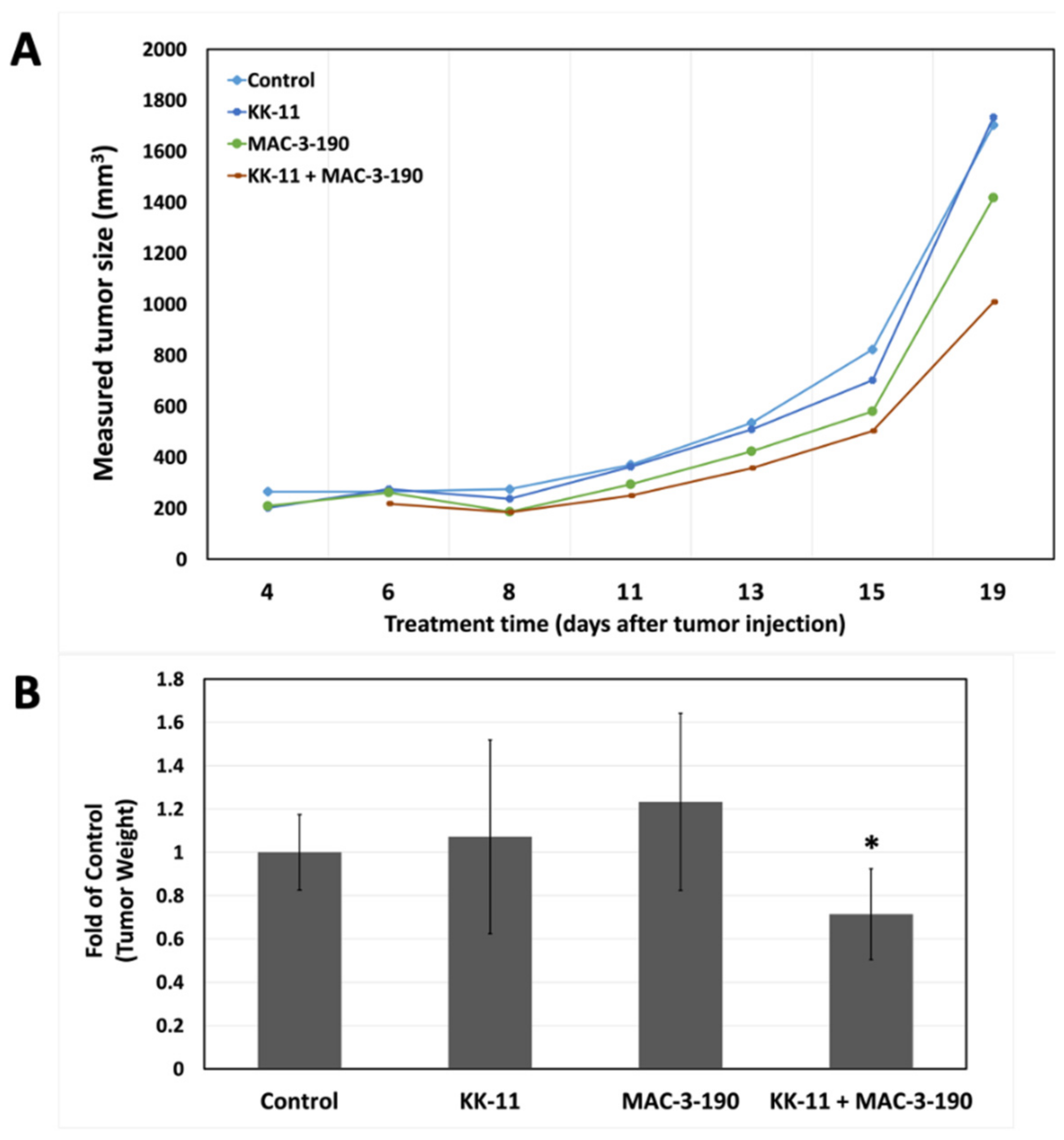
3.5. Bio-Distribution of KK-11 in Tumor Xenografts
3.6. Co-Administration of a Melanoma-Targeted Peptide D-aa KK-11 Enhanced the Antitumor Activity of nNOS Inhibitor MAC-3-190
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- AACR Cancer Progress Report. 2021. Available online: https://cancerprogressreport.aacr.org/wp-content/uploads/sites/2/2021/10/AACR_CPR_2021.pdf (accessed on 10 May 2022).
- Galluzzi, L.; Chan, T.A.; Kroemer, G.; Wolchok, J.D.; Lopez-Soto, A. The hallmarks of successful anticancer immunotherapy. Sci. Transl. Med. 2018, 10, eaat7807. [Google Scholar] [CrossRef] [PubMed]
- Kubli, S.P.; Berger, T.; Araujo, D.V.; Siu, L.L.; Mak, T.W. Beyond immune checkpoint blockade: Emerging immunological strategies. Nat. Rev. Drug Discov. 2021, 20, 899–919. [Google Scholar] [CrossRef] [PubMed]
- Smylie, M.G.; Wong, R.; Mihalcioiu, C.; Lee, C.; Pouliot, J.F. A phase II, open label, monotherapy study of liposomal doxorubicin in patients with metastatic malignant melanoma. Invest. New Drugs 2007, 25, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Fink, W.; Zimpfer-Rechner, C.; Thoelke, A.; Figl, R.; Kaatz, M.; Ugurel, S.; Schadendorf, D. Clinical phase II study of pegylated liposomal doxorubicin as second-line treatment in disseminated melanoma. Onkologie 2004, 27, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Licarete, E.; Rauca, V.F.; Luput, L.; Drotar, D.; Stejerean, I.; Patras, L.; Dume, B.; Toma, V.A.; Porfire, A.; Gherman, C.; et al. Overcoming Intrinsic Doxorubicin Resistance in Melanoma by Anti-Angiogenic and Anti-Metastatic Effects of Liposomal Prednisolone Phosphate on Tumor Microenvironment. Int. J. Mol. Sci. 2020, 21, 2968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheon, S.-H.; Seo, B.-Y.; Lee, Y.-J.; Sim, D.; Lee, S.-B.; Guruprasath, P.; Singh, T.D.; Lee, B.-H.; Sarangthem, V.; Park, R.-W. Targeting of Cisplatin-Resistant Melanoma Using a Multivalent Ligand Presenting an Elastin-like Polypeptide. ACS Biomater. Sci. Eng. 2020, 6, 5024–5031. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Cinelli, M.A.; El-Sayed, N.S.; Huang, H.; Patel, A.; Silverman, R.B.; Yang, S. Inhibition of interferon-gamma-stimulated melanoma progression by targeting neuronal nitric oxide synthase (nNOS). Sci. Rep. 2022, 12, 1701. [Google Scholar] [CrossRef]
- Yang, Z.; Misner, B.; Ji, H.; Poulos, T.L.; Silverman, R.B.; Meyskens, F.L.; Yang, S. Targeting nitric oxide signaling with nNOS inhibitors as a novel strategy for the therapy and prevention of human melanoma. Antioxid. Redox Signal. 2013, 19, 433–447. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Li, H.; Yang, S.; Chreifi, G.; Martasek, P.; Roman, L.J.; Meyskens, F.L.; Poulos, T.L.; Silverman, R.B. Potent and selective double-headed thiophene-2-carboximidamide inhibitors of neuronal nitric oxide synthase for the treatment of melanoma. J. Med. Chem. 2014, 57, 686–700. [Google Scholar] [CrossRef]
- Cinelli, M.A.; Li, H.; Chreifi, G.; Poulos, T.L.; Silverman, R.B. Nitrile in the Hole: Discovery of a Small Auxiliary Pocket in Neuronal Nitric Oxide Synthase Leading to the Development of Potent and Selective 2-Aminoquinoline Inhibitors. J. Med. Chem. 2017, 60, 3958–3978. [Google Scholar] [CrossRef] [Green Version]
- Kalal, B.S.; Upadhya, D.; Pai, V.R. Chemotherapy Resistance Mechanisms in Advanced Skin Cancer. Oncol. Rev. 2017, 11, 326. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Das, M.; Liu, Y.; Huang, L. Targeted drug delivery to melanoma. Adv. Drug Deliv. Rev. 2018, 127, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.G.; Valencia, J.C.; Gillet, J.P.; Hearing, V.J.; Gottesman, M.M. Involvement of ABC transporters in melanogenesis and the development of multidrug resistance of melanoma. Pigment Cell Melanoma Res. 2009, 22, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef]
- Alas, M.; Saghaeidehkordi, A.; Kaur, K. Peptide-Drug Conjugates with Different Linkers for Cancer Therapy. J. Med. Chem. 2021, 64, 216–232. [Google Scholar] [CrossRef] [PubMed]
- Eberle, A.N.; Rout, B.; Qi, M.B.; Bigliardi, P.L. Synthetic Peptide Drugs for Targeting Skin Cancer: Malignant Melanoma and Melanotic Lesions. Curr. Med. Chem. 2017, 24, 1797–1826. [Google Scholar] [CrossRef] [Green Version]
- Redko, B.; Tuchinsky, H.; Segal, T.; Tobi, D.; Luboshits, G.; Ashur-Fabian, O.; Pinhasov, A.; Gerlitz, G.; Gellerman, G. Toward the development of a novel non-RGD cyclic peptide drug conjugate for treatment of human metastatic melanoma. Oncotarget 2017, 8, 757–768. [Google Scholar] [CrossRef] [Green Version]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Greenwald, D.R.; Ruoslahti, E. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science 2010, 328, 1031–1035. [Google Scholar] [CrossRef] [Green Version]
- Barenholz, Y.; Peer, D. Liposomes and other assemblies as drugs and nano-drugs: From basic and translational research to the clinics. J. Control. Release 2012, 160, 115–116. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Mathews, A.S.; Byeon, N.; Lavasanifar, A.; Kaur, K. Peptide arrays for screening cancer specific peptides. Anal. Chem. 2010, 82, 7533–7541. [Google Scholar] [CrossRef]
- Hossein-Nejad-Ariani, H.; Althagafi, E.; Kaur, K. Small Peptide Ligands for Targeting EGFR in Triple Negative Breast Cancer Cells. Sci. Rep. 2019, 9, 2723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, K.M.; Postovit, L.M. Investigating the utility of human melanoma cell lines as tumour models. Oncotarget 2017, 8, 10498–10509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WM3211 Viable Cells. Available online: https://www.rockland.com/categories/cell-lines-and-lysates/wm3211-viable-cells-1-million-cells-WM3211-01-0001/?id=51810 (accessed on 10 May 2022).
- Soudy, R.; Gill, A.; Sprules, T.; Lavasanifar, A.; Kaur, K. Proteolytically stable cancer targeting peptides with high affinity for breast cancer cells. J. Med. Chem. 2011, 54, 7523–7534. [Google Scholar] [CrossRef] [PubMed]
- Raghuwanshi, Y.; Etayash, H.; Soudy, R.; Paiva, I.; Lavasanifar, A.; Kaur, K. Proteolytically Stable Cyclic Decapeptide for Breast Cancer Cell Targeting. J. Med. Chem. 2017, 60, 4893–4903. [Google Scholar] [CrossRef]
- Soudy, R.; Chen, C.; Kaur, K. Novel peptide-doxorubucin conjugates for targeting breast cancer cells including the multidrug resistant cells. J. Med. Chem. 2013, 56, 7564–7573. [Google Scholar] [CrossRef]
- Soudy, R.; Etayash, H.; Bahadorani, K.; Lavasanifar, A.; Kaur, K. Breast Cancer Targeting Peptide Binds Keratin 1: A New Molecular Marker for Targeted Drug Delivery to Breast Cancer. Mol. Pharm. 2017, 14, 593–604. [Google Scholar] [CrossRef]
- Ziaei, E.; Saghaeidehkordi, A.; Dill, C.; Maslennikov, I.; Chen, S.; Kaur, K. Targeting Triple Negative Breast Cancer Cells with Novel Cytotoxic Peptide-Doxorubicin Conjugates. Bioconjug. Chem. 2019, 30, 3098–3106. [Google Scholar] [CrossRef]
- Duong, D.T.; Singh, S.; Bagheri, M.; Verma, N.K.; Schmidtchen, A.; Malmsten, M. Pronounced peptide selectivity for melanoma through tryptophan end-tagging. Sci. Rep. 2016, 6, 24952. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Spring, H.; Schwab, M. Neuroblastoma tumor cell-binding peptides identified through random peptide phage display. Cancer Lett. 2001, 171, 153–164. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, R.; Wu, X.; Sun, Y.; Yao, M.; Li, J.; Xu, Y.; Gu, J. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. FASEB J. 2005, 19, 1978–1985. [Google Scholar] [CrossRef]
- Zhang, L.; Giraudo, E.; Hoffman, J.A.; Hanahan, D.; Ruoslahti, E. Lymphatic zip codes in premalignant lesions and tumors. Cancer Res. 2006, 66, 5696–5706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumthekar, P.; Tang, S.C.; Brenner, A.J.; Kesari, S.; Piccioni, D.E.; Anders, C.; Carrillo, J.; Chalasani, P.; Kabos, P.; Puhalla, S.; et al. ANG1005, a Brain-Penetrating Peptide-Drug Conjugate, Shows Activity in Patients with Breast Cancer with Leptomeningeal Carcinomatosis and Recurrent Brain Metastases. Clin. Cancer Res. 2020, 26, 2789–2799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, A.S.; Chapman, P.B. The history and future of chemotherapy for melanoma. Hematol. Oncol. Clin. N. Am. 2009, 23, 583–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassano, R.; Cuconato, M.; Calviello, G.; Serini, S.; Trombino, S. Recent Advances in Nanotechnology for the Treatment of Melanoma. Molecules 2021, 26, 785. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Tan, S.; Li, S.; Shen, Q.; Wang, K. Cancer drug delivery in the nano era: An overview and perspectives (Review). Oncol. Rep. 2017, 38, 611–624. [Google Scholar] [CrossRef] [Green Version]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody-drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody-drug conjugates for cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef]
- Espelin, C.W.; Leonard, S.C.; Geretti, E.; Wickham, T.J.; Hendriks, B.S. Dual HER2 Targeting with Trastuzumab and Liposomal-Encapsulated Doxorubicin (MM-302) Demonstrates Synergistic Antitumor Activity in Breast and Gastric Cancer. Cancer Res. 2016, 76, 1517–1527. [Google Scholar] [CrossRef] [Green Version]
- Thomas, T.P.; Patri, A.K.; Myc, A.; Myaing, M.T.; Ye, J.Y.; Norris, T.B.; Baker, J.R., Jr. In vitro targeting of synthesized antibody-conjugated dendrimer nanoparticles. Biomacromolecules 2004, 5, 2269–2274. [Google Scholar] [CrossRef]
- Gao, Y.; Zhou, Y.; Zhao, L.; Zhang, C.; Li, Y.; Li, J.; Li, X.; Liu, Y. Enhanced antitumor efficacy by cyclic RGDyK-conjugated and paclitaxel-loaded pH-responsive polymeric micelles. Acta Biomater. 2015, 23, 127–135. [Google Scholar] [CrossRef]
- Froidevaux, S.; Calame-Christe, M.; Tanner, H.; Eberle, A.N. Melanoma targeting with DOTA-alpha-melanocyte-stimulating hormone analogs: Structural parameters affecting tumor uptake and kidney uptake. J. Nucl. Med. 2005, 46, 887–895. [Google Scholar] [PubMed]
- Beckman, R.A.; Weiner, L.M.; Davis, H.M. Antibody constructs in cancer therapy: Protein engineering strategies to improve exposure in solid tumors. Cancer 2007, 109, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Trier, N.; Hansen, P.; Houen, G. Peptides, Antibodies, Peptide Antibodies and More. Int. J. Mol. Sci. 2019, 20, 6289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, F.A.; Stickler, M.M.; Razo, J.; DuBridge, R.B. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the CDR regions. MAbs 2010, 2, 256–265. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.; Teicher, B.A.; Hassan, R. Antibody-drug conjugates for cancer therapy. Lancet Oncol. 2016, 17, e254–e262. [Google Scholar] [CrossRef]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [Green Version]
- Ayo, A.; Laakkonen, P. Peptide-Based Strategies for Targeted Tumor Treatment and Imaging. Pharmaceutics 2021, 13, 481. [Google Scholar] [CrossRef]
- Wang, Y.; Cheetham, A.G.; Angacian, G.; Su, H.; Xie, L.S.; Cui, H.G. Peptide-drug conjugates as effective prodrug strategies for targeted delivery. Adv. Drug Deliver. Rev. 2017, 110, 112–126. [Google Scholar] [CrossRef] [Green Version]
- Apostolopoulos, V.; Bojarska, J.; Chai, T.T.; Elnagdy, S.; Kaczmarek, K.; Matsoukas, J.; New, R.; Parang, K.; Lopez, O.P.; Parhiz, H.; et al. A Global Review on Short Peptides: Frontiers and Perspectives. Molecules 2021, 26, 430. [Google Scholar] [CrossRef]
- Ragozin, E.; Hesin, A.; Bazylevich, A.; Tuchinsky, H.; Bovina, A.; Shekhter Zahavi, T.; Oron-Herman, M.; Kostenich, G.; Firer, M.A.; Rubinek, T.; et al. New somatostatin-drug conjugates for effective targeting pancreatic cancer. Bioorg. Med. Chem. 2018, 26, 3825–3836. [Google Scholar] [CrossRef]
- Ogunnigbagbe, O.; Bunick, C.G.; Kaur, K. Keratin 1 as a cell-surface receptor in cancer. Biochim. Biophys. Acta Rev. Cancer 2021, 1877, 188664. [Google Scholar] [CrossRef] [PubMed]
- Bushnell, D.L.; Bodeker, K.L. Overview and Current Status of Peptide Receptor Radionuclide Therapy. Surg. Oncol. Clin. N. Am. 2020, 29, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Hoppenz, P.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide-Drug Conjugates and Their Targets in Advanced Cancer Therapies. Front. Chem. 2020, 8, 571. [Google Scholar] [CrossRef] [PubMed]
- Vrettos, E.I.; Mezo, G.; Tzakos, A.G. On the design principles of peptide-drug conjugates for targeted drug delivery to the malignant tumor site. Beilstein. J. Org. Chem. 2018, 14, 930–954. [Google Scholar] [CrossRef]
- Kristensen, M.; Franzyk, H.; Klausen, M.T.; Iversen, A.; Bahnsen, J.S.; Skyggebjerg, R.B.; Fodera, V.; Nielsen, H.M. Penetratin-Mediated Transepithelial Insulin Permeation: Importance of Cationic Residues and pH for Complexation and Permeation. AAPS J. 2015, 17, 1200–1209. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, M.; Birch, D.; Morck Nielsen, H. Applications and Challenges for Use of Cell-Penetrating Peptides as Delivery Vectors for Peptide and Protein Cargos. Int. J. Mol. Sci. 2016, 17, 185. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Takahashi, T. Pathophysiological significance of neuronal nitric oxide synthase in the gastrointestinal tract. J. Gastroenterol. 2003, 38, 421–430. [Google Scholar] [CrossRef]
- Orihata, M.; Sarna, S.K. Inhibition of nitric oxide synthase delays gastric emptying of solid meals. J. Pharmacol. Exp. Ther. 1994, 271, 660–670. [Google Scholar]
- Caputi, V.; Marsilio, I.; Filpa, V.; Cerantola, S.; Orso, G.; Bistoletti, M.; Paccagnella, N.; De Martin, S.; Montopoli, M.; Dall’Acqua, S.; et al. Antibiotic-induced dysbiosis of the microbiota impairs gut neuromuscular function in juvenile mice. Br. J. Pharmacol. 2017, 174, 3623–3639. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.L.; Dawson, T.M.; Bredt, D.S.; Snyder, S.H.; Fishman, M.C. Targeted disruption of the neuronal nitric oxide synthase gene. Cell 1993, 75, 1273–1286. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Number | Amino Acid Sequence | Fluorescence Ratio (Melanoma/Non-Melanoma Cells) * | Peptide Number | Amino Acid Sequence | Fluorescence Ratio (Melanoma/Non-Melanoma Cells) * |
|---|---|---|---|---|---|
| 1 | GRRPRPRPRP | 1.82 | 28 | YHWYGYAPQNVI | 1.26 |
| 2 | GRRPRPRPRPW | 1.60 | 29 | YHWYGYTAQNVI | 1.32 |
| 3 | GRRPRPRPRPWW | 1.50 | 30 | YHWYGYTPANVI | 1.30 |
| 4 | GRRPRPRPRPWWW | 1.46 | 31 | YHWYGYTPQAVI | 1.33 |
| 5 | GRRPRPRPRPWWWW | 1.43 | 32 | YHWYGYTPQNAI | 1.29 |
| 6 | GARPRPRPRP | 1.37 | 33 | YHWYGYTPQNVA | 1.35 |
| 7 | GRAPRPRPRP | 1.47 | 34 | YHWYGYTPENVI | 1.36 |
| 8 | GRRPAPRPRP | 1.57 | 35 | YHWYGYTPQDVI | 1.35 |
| 9 | GRRPRPAPRP | 1.51 | 36 | YHWYGYTPQKVI | 1.23 |
| 10 | GRRPRPRPAP | 1.31 | 37 | CLSDGKRKC | 1.29 |
| 11 | VPWXEPAYQRFL | 1.80 | 38 | AGRKLDSKA | 1.39 |
| 12 | WXEAAYQRFL | 1.31 | 39 | ADRSKGKLA | 1.27 |
| 13 | EPAAYQRFTA | 1.39 | 40 | ALSDGKRKA | 1.23 |
| 14 | RVPWLEAPYA | 1.42 | 41 | ALSDGKRKC | 1.32 |
| 15 | FVPWLEAPYA | 1.61 | 42 | CASDGKRKC | 1.25 |
| 16 | RGDAAYQRFL | 1.47 | 43 | CLADGKRKC | 1.26 |
| 17 | RGEPAYQRFL | 1.30 | 44 | CLSAGKRKC | 1.44 |
| 18 | RGEPAYQGRFL | 1.24 | 45 | CLSDAKRKC | 1.22 |
| 19 | RGDPAYQGRFL | 1.35 | 46 | CLSDGARKC | 1.35 |
| 20 | YHWYGYTPQNVI | 1.20 | 47 | CLSDGKAKC | 1.27 |
| 21 | WQTNYIHPYVYG | 1.21 | 48 | CLSDGKRAC | 1.12 |
| 22 | YGPWYNHYITQV | 1.28 | 49 | CLSDGKRKA | 1.57 |
| 23 | AHWYGYTPQNVI | 1.31 | 50 | CLSEGKRKC | 1.38 |
| 24 | YAWYGYTPQNVI | 1.15 | 51 | CLSDGRRKC | 1.32 |
| 25 | YHAYGYTPQNVI | 1.28 | 52 | CLSDGKRRC | 1.27 |
| 26 | YHWAGYTPQNVI | 1.29 | 53 | WLSDGKRKC | 1.12 |
| 27 | YHWYGATPQNVI | 1.19 |
| Peptide | Label | [M + H]+ | |
|---|---|---|---|
| Calculated | Found | ||
| VPWXEPAYQRFL | KK-11 | 1517.8 | 1518.1 |
| VPWxEPAYQrFL | D-aa KK-11 | 1517.8 | 1517.6 |
| FITC-Aβ-VPWXEPAYQRFL | FITC-KK-11 | 1979.2 | 1979.1 |
| VivoTag-Aβ-VPWxEPAYQrFL | VivoTag-KK-11 | 2823.2 | 2823.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tong, S.; Darwish, S.; Ariani, H.H.N.; Lozada, K.A.; Salehi, D.; Cinelli, M.A.; Silverman, R.B.; Kaur, K.; Yang, S. A Small Peptide Increases Drug Delivery in Human Melanoma Cells. Pharmaceutics 2022, 14, 1036. https://doi.org/10.3390/pharmaceutics14051036
Tong S, Darwish S, Ariani HHN, Lozada KA, Salehi D, Cinelli MA, Silverman RB, Kaur K, Yang S. A Small Peptide Increases Drug Delivery in Human Melanoma Cells. Pharmaceutics. 2022; 14(5):1036. https://doi.org/10.3390/pharmaceutics14051036
Chicago/Turabian StyleTong, Shirley, Shaban Darwish, Hanieh Hossein Nejad Ariani, Kate Alison Lozada, David Salehi, Maris A. Cinelli, Richard B. Silverman, Kamaljit Kaur, and Sun Yang. 2022. "A Small Peptide Increases Drug Delivery in Human Melanoma Cells" Pharmaceutics 14, no. 5: 1036. https://doi.org/10.3390/pharmaceutics14051036