
Identification of Somatic Mitochondrial DNA Mutations, Heteroplasmy, and Increased Levels of Catenanes in Tumor Specimens Obtained from Three Endometrial Cancer Patients

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Tissue Specimens
2.2. DNA Extraction from Normal and Tumor Tissue
2.3. mtDNA Next-Generation Sequencing and Data Analysis
2.4. Determination of Relative mtDNA Copy Number
2.5. Characterization of mtDNA Topoisomers
3. Results
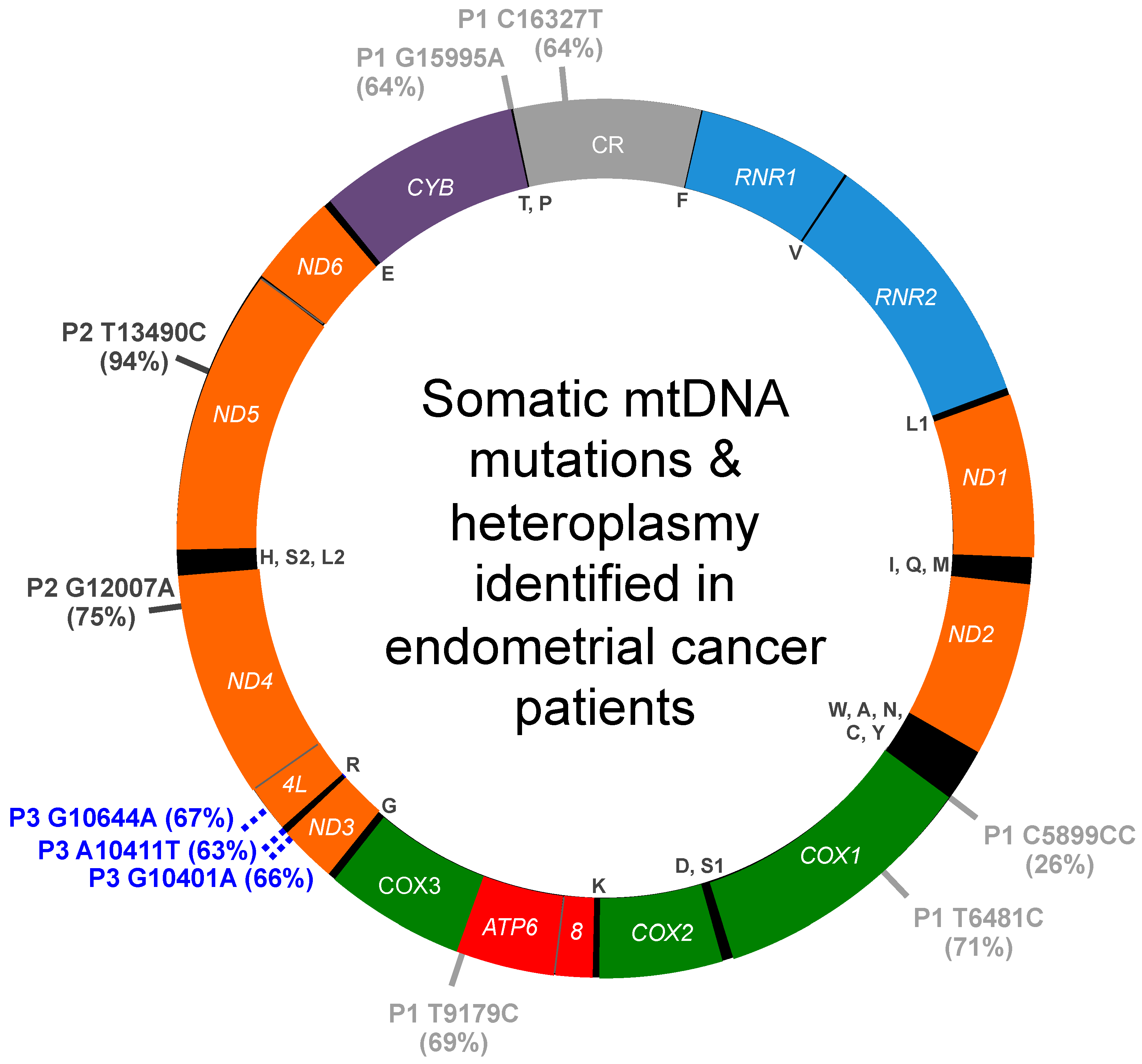
3.1. mtDNA Mutations and Heteroplasmy Identified in Patient 1
3.2. mtDNA Mutations and Heteroplasmy Identified in Patient 2
3.3. mtDNA Mutations and Heteroplasmy Identified in Patient 3
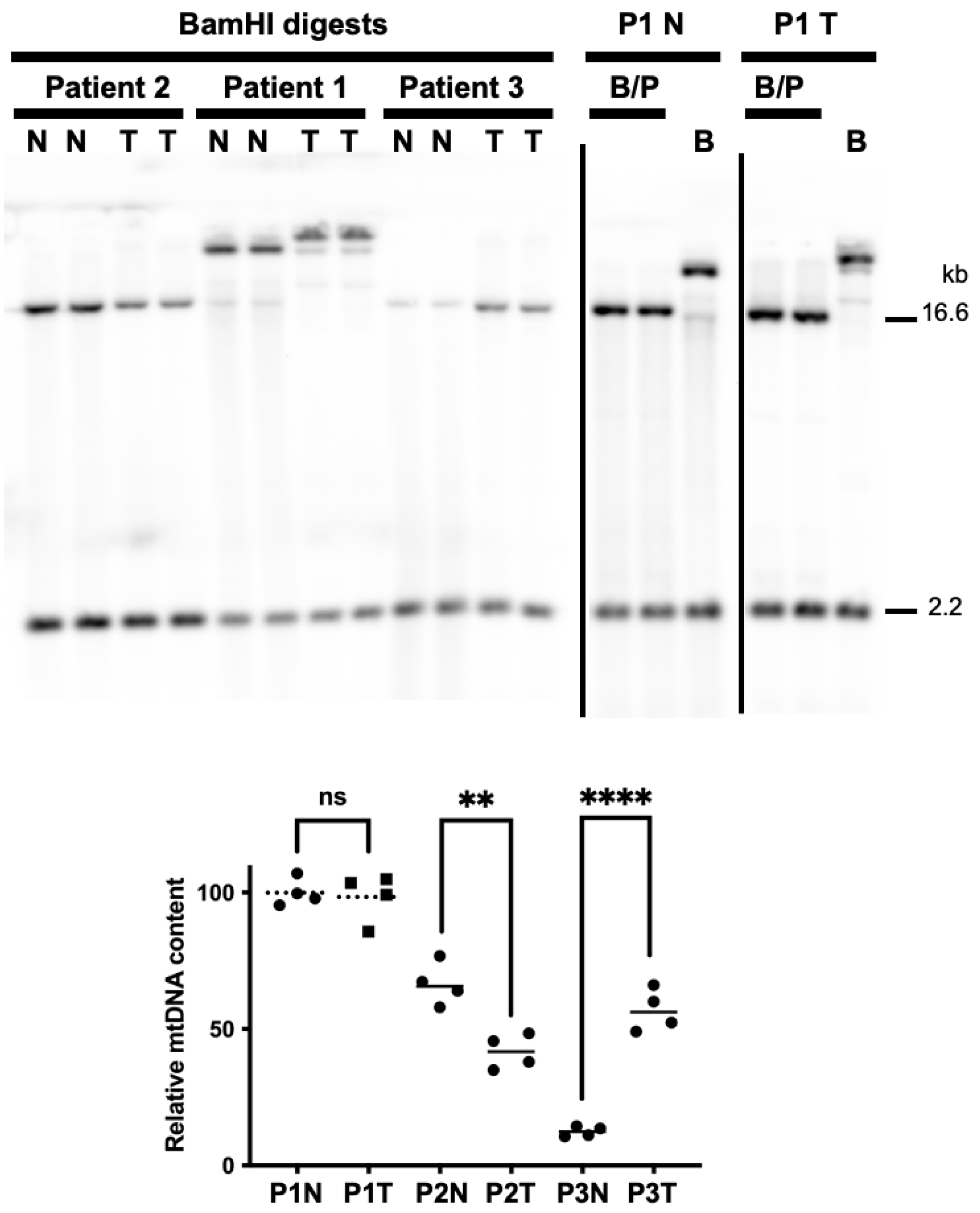
3.4. Tumor DNA Extracts from Patients 2 and 3 Harbor Altered mtDNA Copy Number
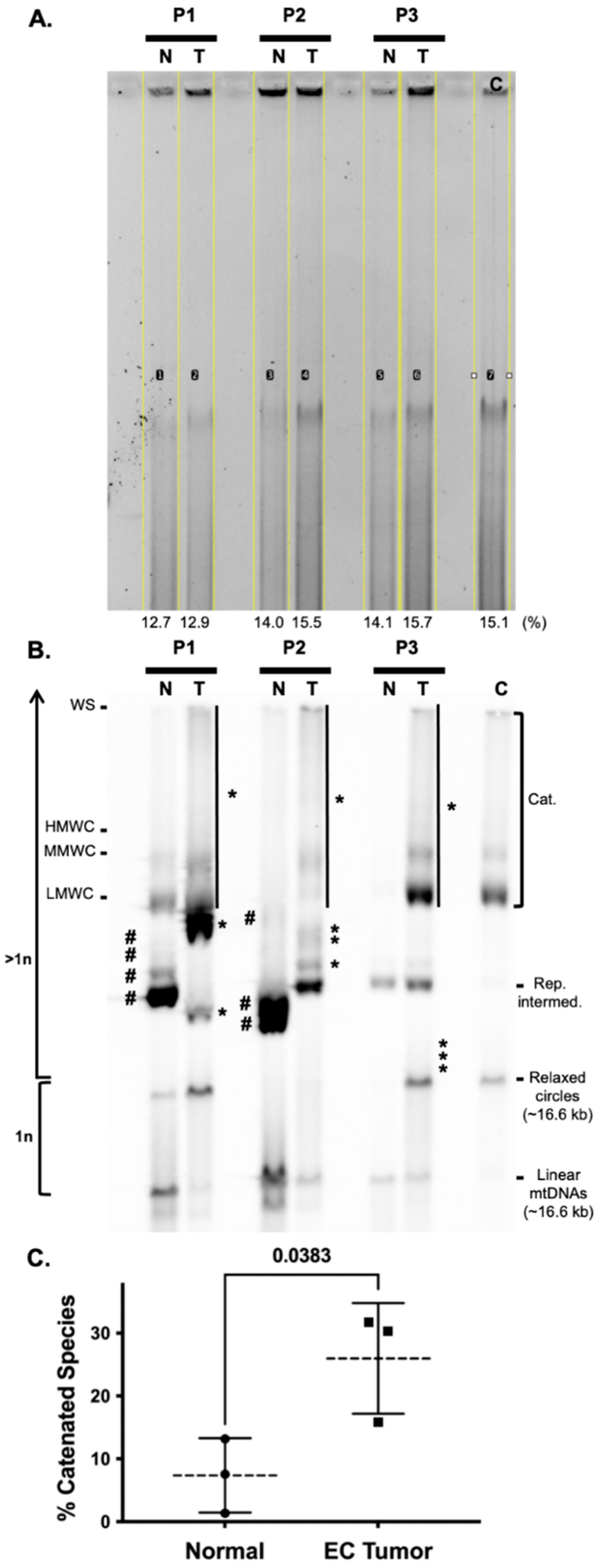
3.5. Endometrial Cancer Tumors Have Striking Differences in mtDNA Topoisomer Molecules
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braun, M.M.; Overbeek-Wager, E.A.; Grumbo, R.J. Diagnosis and Management of Endometrial Cancer. Am. Fam. Physician 2016, 93, 468–474. [Google Scholar] [PubMed]
- Zhang, S.; Gong, T.T.; Liu, F.H.; Jiang, Y.T.; Sun, H.; Ma, X.X.; Zhao, Y.H.; Wu, Q.J. Global, Regional, and National Burden of Endometrial Cancer, 1990-2017: Results From the Global Burden of Disease Study, 2017. Front. Oncol. 2019, 9, 1440. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, A.K.; Buck, V.U.; Classen-Linke, I.; Leube, R.E. How Mechanical Forces Change the Human Endometrium during the Menstrual Cycle in Preparation for Embryo Implantation. Cells 2021, 10, 2008. [Google Scholar] [CrossRef] [PubMed]
- Stewart, B.W.; Wild, C. International Agency for Research on Cancer, World Health Organization. In World Cancer Report 2014; International Agency for Research on Cancer, WHO Press: Lyon, France; Geneva, Switzerland, 2014; Volume xiv, p. 630. [Google Scholar]
- Zahnd, W.E.; Hyon, K.S.; Diaz-Sylvester, P.; Izadi, S.R.; Colditz, G.A.; Brard, L. Rural-urban differences in surgical treatment, regional lymph node examination, and survival in endometrial cancer patients. Cancer Causes Control. 2018, 29, 221–232. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Guerra, F.; Kurelac, I.; Cormio, A.; Zuntini, R.; Amato, L.B.; Ceccarelli, C.; Santini, D.; Cormio, G.; Fracasso, F.; Selvaggi, L.; et al. Placing mitochondrial DNA mutations within the progression model of type I endometrial carcinoma. Hum. Mol. Genet 2011, 20, 2394–2405. [Google Scholar] [CrossRef]
- Musicco, C.; Cormio, G.; Pesce, V.; Loizzi, V.; Cicinelli, E.; Resta, L.; Ranieri, G.; Cormio, A. Mitochondrial Dysfunctions in Type I Endometrial Carcinoma: Exploring Their Role in Oncogenesis and Tumor Progression. Int. J. Mol. Sci. 2018, 19, 2076. [Google Scholar] [CrossRef] [Green Version]
- Chang, M. Dual roles of estrogen metabolism in mammary carcinogenesis. BMB Rep. 2011, 44, 423–434. [Google Scholar] [CrossRef]
- Gammage, P.A.; Frezza, C. Mitochondrial DNA: The overlooked oncogenome? BMC Biol. 2019, 17, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martincorena, I.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. Elife 2014, 3, e02935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, J.B.; Alaei-Mahabadi, B.; Sabarinathan, R.; Samuelsson, T.; Gorodkin, J.; Gustafsson, C.M.; Larsson, E. Simultaneous DNA and RNA Mapping of Somatic Mitochondrial Mutations across Diverse Human Cancers. PLoS Genet 2015, 11, e1005333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schon, E.A.; DiMauro, S.; Hirano, M. Human mitochondrial DNA: Roles of inherited and somatic mutations. Nat. Rev. Genet 2012, 13, 878–890. [Google Scholar] [CrossRef]
- Santos, J.H.; Hunakova, L.; Chen, Y.; Bortner, C.; Van Houten, B. Cell sorting experiments link persistent mitochondrial DNA damage with loss of mitochondrial membrane potential and apoptotic cell death. J. Biol. Chem. 2003, 278, 1728–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.J.; Copeland, W.C. Human mitochondrial DNA replication machinery and disease. Curr. Opin. Genet Dev. 2016, 38, 52–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.J.; Humble, M.M.; DeBalsi, K.L.; Sun, K.Y.; Copeland, W.C. POLG2 disease variants: Analyses reveal a dominant negative heterodimer, altered mitochondrial localization and impaired respiratory capacity. Hum. Mol. Genet 2015, 24, 5184–5197. [Google Scholar] [CrossRef] [Green Version]
- Jayaprakash, A.D.; Benson, E.K.; Gone, S.; Liang, R.; Shim, J.; Lambertini, L.; Toloue, M.M.; Wigler, M.; Aaronson, S.A.; Sachidanandam, R. Stable heteroplasmy at the single-cell level is facilitated by intercellular exchange of mtDNA. Nucleic Acids Res. 2015, 43, 2177–2187. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Wu, J.; Dressman, D.C.; Iacobuzio-Donahue, C.; Markowitz, S.D.; Velculescu, V.E.; Diaz, L.A., Jr.; Kinzler, K.W.; Vogelstein, B.; Papadopoulos, N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature 2010, 464, 610–614. [Google Scholar] [CrossRef] [Green Version]
- Bayona-Bafaluy, M.P.; Manfredi, G.; Moraes, C.T. A chemical enucleation method for the transfer of mitochondrial DNA to rho(o) cells. Nucleic Acids Res. 2003, 31, e98. [Google Scholar] [CrossRef] [Green Version]
- King, M.P.; Attardi, G. Isolation of human cell lines lacking mitochondrial DNA. Methods Enzymol. 1996, 264, 304–313. [Google Scholar] [PubMed]
- Carelli, V.; Vergani, L.; Bernazzi, B.; Zampieron, C.; Bucchi, L.; Valentino, M.; Rengo, C.; Torroni, A.; Martinuzzi, A. Respiratory function in cybrid cell lines carrying European mtDNA haplogroups: Implications for Leber’s hereditary optic neuropathy. Biochim. Biophys. Acta 2002, 1588, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Heller, S.; Schubert, S.; Krehan, M.; Schafer, I.; Seibel, M.; Latorre, D.; Villani, G.; Seibel, P. Efficient repopulation of genetically derived rho zero cells with exogenous mitochondria. PLoS ONE 2013, 8, e73207. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.Q.; Henning, M.S.; Starkov, A.A.; Manfredi, G. The mitochondrial respiratory chain is a modulator of apoptosis. J. Cell Biol. 2007, 179, 1163–1177. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [Green Version]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D’Agostino, D.P. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis 2014, 35, 515–527. [Google Scholar] [CrossRef]
- Gaude, E.; Schmidt, C.; Gammage, P.A.; Dugourd, A.; Blacker, T.; Chew, S.P.; Saez-Rodriguez, J.; O’Neill, J.S.; Szabadkai, G.; Minczuk, M.; et al. NADH Shuttling Couples Cytosolic Reductive Carboxylation of Glutamine with Glycolysis in Cells with Mitochondrial Dysfunction. Mol. Cell 2018, 69, 581–593.e587. [Google Scholar] [CrossRef] [Green Version]
- Young, M.J.; Jayaprakash, A.D.; Young, C.K.J. Analysis of Mitochondrial DNA Polymorphisms in the Human Cell Lines HepaRG and SJCRH30. Int. J. Mol. Sci. 2019, 20, 3245. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C.; Stugard, C.; Murdock, D.; Schurr, T.; Brown, M.D. Ancient mtDNA sequences in the human nuclear genome: A potential source of errors in identifying pathogenic mutations. Proc. Natl. Acad. Sci. USA 1997, 94, 14900–14905. [Google Scholar] [CrossRef] [Green Version]
- Grandhi, S.; Bosworth, C.; Maddox, W.; Sensiba, C.; Akhavanfard, S.; Ni, Y.; LaFramboise, T. Heteroplasmic shifts in tumor mitochondrial genomes reveal tissue-specific signals of relaxed and positive selection. Hum. Mol. Genet 2017, 26, 2912–2922. [Google Scholar] [CrossRef]
- Young, C.K.J.; Wheeler, J.H.; Rahman, M.M.; Young, M.J. The antiretroviral 2′,3′-dideoxycytidine causes mitochondrial dysfunction in proliferating and differentiated HepaRG human cell cultures. J. Biol. Chem. 2021, 296, 100206. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Ju, Y.S.; Kim, Y.; Li, J.; Wang, Y.; Yoon, C.J.; Yang, Y.; Martincorena, I.; Creighton, C.J.; Weinstein, J.N.; et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat. Genet. 2020, 52, 342–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, J.H.; Young, C.K.J.; Young, M.J. Analysis of Human Mitochondrial DNA Content by Southern Blotting and Nonradioactive Probe Hybridization. Curr. Protoc. Toxicol. 2019, 80, e75. [Google Scholar] [CrossRef] [PubMed]
- Lott, M.T.; Leipzig, J.N.; Derbeneva, O.; Xie, H.M.; Chalkia, D.; Sarmady, M.; Procaccio, V.; Wallace, D.C. mtDNA Variation and Analysis Using Mitomap and Mitomaster. Curr. Protoc. Bioinform. 2013, 44, 21–26. [Google Scholar] [CrossRef] [Green Version]
- Castellana, S.; Fusilli, C.; Mazzoccoli, G.; Biagini, T.; Capocefalo, D.; Carella, M.; Vescovi, A.L.; Mazza, T. High-confidence assessment of functional impact of human mitochondrial non-synonymous genome variations by APOGEE. PLoS Comput. Biol. 2017, 13, e1005628. [Google Scholar] [CrossRef]
- Castellana, S.; Biagini, T.; Petrizzelli, F.; Parca, L.; Panzironi, N.; Caputo, V.; Vescovi, A.L.; Carella, M.; Mazza, T. MitImpact 3: Modeling the residue interaction network of the Respiratory Chain subunits. Nucleic Acids Res. 2021, 49, D1282–D1288. [Google Scholar] [CrossRef]
- Sonney, S.; Leipzig, J.; Lott, M.T.; Zhang, S.; Procaccio, V.; Wallace, D.C.; Sondheimer, N. Predicting the pathogenicity of novel variants in mitochondrial tRNA with MitoTIP. PLoS Comput. Biol. 2017, 13, e1005867. [Google Scholar] [CrossRef]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [Green Version]
- Herrnstadt, C.; Preston, G.; Andrews, R.; Chinnery, P.; Lightowlers, R.N.; Turnbull, D.M.; Kubacka, I.; Howell, N. A high frequency of mtDNA polymorphisms in HeLa cell sublines. Mutat. Res. 2002, 501, 19–28. [Google Scholar] [CrossRef]
- Wong, L.J.; Liang, M.H.; Kwon, H.; Park, J.; Bai, R.K.; Tan, D.J. Comprehensive scanning of the entire mitochondrial genome for mutations. Clin. Chem. 2002, 48, 1901–1912. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Zhai, K.; Chang, L.; Zhang, Q.; Liu, B.; Wu, Y. Mitochondrial C150T polymorphism increases the risk of cervical cancer and HPV infection. Mitochondrion 2011, 11, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, D.; Bai, R.K.; Wong, L.C.; Leal, S.M. Mitochondrial DNA variant interactions modify breast cancer risk. J. Hum. Genet. 2008, 53, 924–928. [Google Scholar] [CrossRef] [Green Version]
- Booker, L.M.; Habermacher, G.M.; Jessie, B.C.; Sun, Q.C.; Baumann, A.K.; Amin, M.; Lim, S.D.; Fernandez-Golarz, C.; Lyles, R.H.; Brown, M.D.; et al. North American white mitochondrial haplogroups in prostate and renal cancer. J. Urol. 2006, 175, 468–472; discussion 463–472. [Google Scholar] [CrossRef]
- Ebner, S.; Lang, R.; Mueller, E.E.; Eder, W.; Oeller, M.; Moser, A.; Koller, J.; Paulweber, B.; Mayr, J.A.; Sperl, W.; et al. Mitochondrial haplogroups, control region polymorphisms and malignant melanoma: A study in middle European Caucasians. PLoS ONE 2011, 6, e27192. [Google Scholar] [CrossRef]
- Lauber, J.; Marsac, C.; Kadenbach, B.; Seibel, P. Mutations in mitochondrial tRNA genes: A frequent cause of neuromuscular diseases. Nucleic Acids Res. 1991, 19, 1393–1397. [Google Scholar] [CrossRef]
- Pulkes, T.; Sweeney, M.G.; Hanna, M.G. Increased risk of stroke in patients with the A12308G polymorphism in mitochondria. Lancet 2000, 356, 2068–2069. [Google Scholar] [CrossRef]
- Merante, F.; Tein, I.; Benson, L.; Robinson, B.H. Maternally inherited hypertrophic cardiomyopathy due to a novel T-to-C transition at nucleotide 9997 in the mitochondrial tRNA(glycine) gene. Am. J. Hum. Genet. 1994, 55, 437–446. [Google Scholar]
- Zhang, J.; Zhang, Z.X.; Du, P.C.; Zhou, W.; Wu, S.D.; Wang, Q.L.; Chen, C.; Shi, Q.; Chen, C.; Gao, C.; et al. Analyses of the mitochondrial mutations in the Chinese patients with sporadic Creutzfeldt-Jakob disease. Eur. J. Hum. Genet. 2015, 23, 86–91. [Google Scholar] [CrossRef] [Green Version]
- Rollins, B.; Martin, M.V.; Sequeira, P.A.; Moon, E.A.; Morgan, L.Z.; Watson, S.J.; Schatzberg, A.; Akil, H.; Myers, R.M.; Jones, E.G.; et al. Mitochondrial variants in schizophrenia, bipolar disorder, and major depressive disorder. PLoS ONE 2009, 4, e4913. [Google Scholar] [CrossRef] [PubMed]
- Aitullina, A.; Baumane, K.; Zalite, S.; Ranka, R.; Zole, E.; Pole, I.; Sepetiene, S.; Laganovska, G.; Baumanis, V.; Pliss, L. Point mutations associated with Leber hereditary optic neuropathy in a Latvian population. Mol. Vis. 2013, 19, 2343–2351. [Google Scholar] [PubMed]
- Sequeira, A.; Martin, M.V.; Rollins, B.; Moon, E.A.; Bunney, W.E.; Macciardi, F.; Lupoli, S.; Smith, E.N.; Kelsoe, J.; Magnan, C.N.; et al. Mitochondrial mutations and polymorphisms in psychiatric disorders. Front. Genet. 2012, 3, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.Y.; Maisch, B.; Rose, M.L.; Hengstenberg, C. Point mutations in mitochondrial DNA of patients with dilated cardiomyopathy. J. Mol. Cell Cardiol. 1997, 29, 2699–2709. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, M.; Gustafsson, C.M. Mammalian mitochondrial DNA replication and mechanisms of deletion formation. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 509–524. [Google Scholar] [CrossRef]
- Gustafsson, C.M.; Falkenberg, M.; Larsson, N.G. Maintenance and Expression of Mammalian Mitochondrial DNA. Annu. Rev. Biochem. 2016, 85, 133–160. [Google Scholar] [CrossRef]
- Shadel, G.S.; Clayton, D.A. Mitochondrial DNA maintenance in vertebrates. Annu. Rev. Biochem. 1997, 66, 409–435. [Google Scholar] [CrossRef]
- Kolesar, J.E.; Wang, C.Y.; Taguchi, Y.V.; Chou, S.H.; Kaufman, B.A. Two-dimensional intact mitochondrial DNA agarose electrophoresis reveals the structural complexity of the mammalian mitochondrial genome. Nucleic Acids Res. 2013, 41, e58. [Google Scholar] [CrossRef] [Green Version]
- Pohjoismaki, J.L.; Goffart, S.; Tyynismaa, H.; Willcox, S.; Ide, T.; Kang, D.; Suomalainen, A.; Karhunen, P.J.; Griffith, J.D.; Holt, I.J.; et al. Human heart mitochondrial DNA is organized in complex catenated networks containing abundant four-way junctions and replication forks. J. Biol. Chem. 2009, 284, 21446–21457. [Google Scholar] [CrossRef] [Green Version]
- Pohjoismaki, J.L.; Wanrooij, S.; Hyvarinen, A.K.; Goffart, S.; Holt, I.J.; Spelbrink, J.N.; Jacobs, H.T. Alterations to the expression level of mitochondrial transcription factor A, TFAM, modify the mode of mitochondrial DNA replication in cultured human cells. Nucleic Acids Res. 2006, 34, 5815–5828. [Google Scholar] [CrossRef]
- Goffart, S.; Hangas, A.; Pohjoismaki, J.L.O. Twist and Turn-Topoisomerase Functions in Mitochondrial DNA Maintenance. Int. J. Mol. Sci. 2019, 20, 2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avital, G.; Buchshtav, M.; Zhidkov, I.; Tuval Feder, J.; Dadon, S.; Rubin, E.; Glass, D.; Spector, T.D.; Mishmar, D. Mitochondrial DNA heteroplasmy in diabetes and normal adults: Role of acquired and inherited mutational patterns in twins. Hum. Mol. Genet. 2012, 21, 4214–4224. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Soares, P.; Radivojac, P.; Li, B.; Samuels, D.C. Comparing phylogeny and the predicted pathogenicity of protein variations reveals equal purifying selection across the global human mtDNA diversity. Am. J. Hum. Genet. 2011, 88, 433–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Rothwell, R.; Vermaat, M.; Wachsmuth, M.; Schroder, R.; Laros, J.F.; van Oven, M.; de Bakker, P.I.; Bovenberg, J.A.; van Duijn, C.M.; et al. Transmission of human mtDNA heteroplasmy in the Genome of the Netherlands families: Support for a variable-size bottleneck. Genome Res. 2016, 26, 417–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohjoismaki, J.L.; Goffart, S.; Taylor, R.W.; Turnbull, D.M.; Suomalainen, A.; Jacobs, H.T.; Karhunen, P.J. Developmental and pathological changes in the human cardiac muscle mitochondrial DNA organization, replication and copy number. PLoS ONE 2010, 5, e10426. [Google Scholar] [CrossRef] [PubMed]
- Peeva, V.; Blei, D.; Trombly, G.; Corsi, S.; Szukszto, M.J.; Rebelo-Guiomar, P.; Gammage, P.A.; Kudin, A.P.; Becker, C.; Altmuller, J.; et al. Linear mitochondrial DNA is rapidly degraded by components of the replication machinery. Nat. Commun. 2018, 9, 1727. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, M.W.; Kennedy, S.R.; Salk, J.J.; Fox, E.J.; Hiatt, J.B.; Loeb, L.A. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 14508–14513. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Pursell, Z.F.; Copeland, W.C.; Longley, M.J.; Kunkel, T.A.; Mathews, C.K. DNA percursor asymmetries in mammalian tissue mitochondrial and possible contribution to mitochondrial mutagenesis through reduced replication fidleity. Proc. Natl. Acad. Sci. USA 2005, 102, 4990–4995. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, T.J.; Minczuk, M. In D-loop: 40 years of mitochondrial 7S DNA. Exp. Gerontol. 2014, 56, 175–181. [Google Scholar] [CrossRef]
- Mi, J.; Tian, G.; Liu, S.; Li, X.; Ni, T.; Zhang, L.; Wang, B. The relationship between altered mitochondrial DNA copy number and cancer risk: A meta-analysis. Sci. Rep. 2015, 5, 10039. [Google Scholar] [CrossRef] [Green Version]
- Gorelick, A.N.; Kim, M.; Chatila, W.K.; La, K.; Hakimi, A.A.; Berger, M.F.; Taylor, B.S.; Gammage, P.A.; Reznik, E. Respiratory complex and tissue lineage drive recurrent mutations in tumour mtDNA. Nat. Metab. 2021, 3, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Huijgens, A.N.; Mertens, H.J. Factors predicting recurrent endometrial cancer. Facts Views Vis. Obgyn. 2013, 5, 179–186. [Google Scholar] [PubMed]
- Hardarson, H.A.; Heidemann, L.N.; dePont Christensen, R.; Mogensen, O.; Jochumsen, K.M. Vaginal vault recurrences of endometrial cancer in non-irradiated patients-Radiotherapy or surgery. Gynecol. Oncol. Rep. 2015, 11, 26–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutt, S.; Tailor, A.; Ellis, P.; Michael, A.; Butler-Manuel, S.; Chatterjee, J. The role of biomarkers in endometrial cancer and hyperplasia: A literature review. Acta Oncol. 2019, 58, 342–352. [Google Scholar] [CrossRef] [Green Version]
- Rosa, H.S.; Ajaz, S.; Gnudi, L.; Malik, A.N. A case for measuring both cellular and cell-free mitochondrial DNA as a disease biomarker in human blood. FASEB J. 2020, 34, 12278–12288. [Google Scholar] [CrossRef]
- Afrifa, J.; Zhao, T.; Yu, J. Circulating mitochondria DNA, a non-invasive cancer diagnostic biomarker candidate. Mitochondrion 2019, 47, 238–243. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| MtDNA nt change a | Location b | Coverage c | %Variant d | Remarks e |
|---|---|---|---|---|
| C5899CC | NC5, adjacent OL and TRNY | 714 | 26 | Insertion of C, potential effect on DNA rep. initiation? |
| T6481C | COX1/V193A | 794 | 71 | Missense variant, deleterious (1.0) f; pathogenic (0.56) g; functional impact medium (2.34) h; associated with LAML, BRCA/Breast AdenoCA [31,33] |
| T9179C | ATP6/V218A | 767 | 69 | Missense variant, deleterious (0.99) f; neutral (0.4) g; functional impact medium (2.91) h; associated with CLL, Panc-AdenoCA [33] |
| G15995A | TRNP | 756 | 64 | Likely pathogenic (MitoTIP80%) i, mitochondrial cytopathy [41]; associated with BRCA/Breast-AdenoCA [31,33] |
| C16327T | ATT, CR:HVS1,7S | 489 | 64 | Potential effect on DNA rep. initiation? associated with STAD, CLL, PRAD, Panc-AdenoCA [31,33] |
| MtDNA nt Change a | Location | Coverage | %Variant | Remarks |
|---|---|---|---|---|
| G12007A | ND4/W416W | 424 | 75 | Synonymous variant; significantly associated with SZ + BD b [54]; associated with PRAD [33] |
| T13490C | ND5/F385S | 516 | 94 | Missense variant, deleterious (1.0) c; pathogenic (0.63) d; Functional impact medium (3.425) e; associated with CLL, RCC [33] |
| MtDNA nt Change a | Location b | Coverage | %Variant | Remarks |
|---|---|---|---|---|
| G10401A | ND3/E115K | 556 | 66 | Missense variant, deleterious (0.99) c; neutral (0.31) d; associated with THCA, HCC, Ovary-AdenoCA [31,33] |
| A10411T | TRNR | 540 | 63 | Assoc. w/cardiomyopathy [55]; possibly benign(MitoTIP26.4%) e |
| G10644A | ND4L/V59M | 658 | 67 | Missense variant, neutral (0.45) c; neutral (0.3) d; conflicting interpretations of pathogenicity (NC_012920.1(MT-ND4L):m.10644G>A) f; associated with PRDA [33] |
| Sample Index (See Supp. File 2 in [13]) | MtDNA nt Change a | Location b | %Variant | Remarks c |
|---|---|---|---|---|
| 5128 | T5506C | ND2/I346T | 91 | Missense, neutral (0.77) d; neutral (0.45) e; Uterus-AdenoCA [33] |
| 5139 | T11875C | ND4/T372T | 92 | Synonymous, UCEC, Eso-AdenoCA [31,33] |
| 5139 | G13531A | ND5/A399T | 93 | Missense, neutral (0.52) d; neutral (0.29) e; UCEC, Panc-Endorine, Soft Tissue-Liposarc, HCC [31,33] |
| 5150 | G4308A | TRNI | 46 | Confirmed pathogenic (MitoTIP82.1%) f, UCEC, Breast AdenoCA, Kidney-ChRCC [31,33] |
| 5155 | G14410A | ND6/V88V | 35 | Synonymous, UCEC [31] |
| 5157 | C14112T | ND5/F592F | 96 | Synonymous, UCEC/Uterus-AdenoCA [31,33] |
| 5160 | T152C | ATT, CR: HVS2, 7S, OH | 88 | LGG, LUAD/ Lung-AdenoCA, SARC, SKCM, THCA, BLCA/ Bladder-TCC, BRCA/Breast-AdenoCA, ESCA, PRAD/Prost-AdenoCA, STAD, UCEC, Lymph-CLL, Panc-AdenoCA, Soft Tissue-Liposarc, skin melanoma [31,33] |
| 5160 | G1642A | TRNV | 80 | Possibly pathogenic (MitoTIP74.3%) f, HNSC, UCEC, Head-SCC, Panc-AdenoCA, Thy-AdenoCA, HCC [31,33] |
| 5160 | G13417A | ND5/G361TERM | 51 | Nonsense mutation, LUAD, UCEC, HNSC [31] |
| 5161 | G5560A | TRNW | 12 | Possibly pathogenic (MitoTIP62.3%) f, THCA/Thy-AdenoCA, Kidney-ChRCC, Ovary-AdenoCA [31,33] |
| 5163 | T9062C | ATP6/L179P | 14 | Missense, neutral (0.92) d; neutral (0.25) e; UCEC/Uterus-AdenoCA [31,33] |
| 5165 | G529A | CR: HVS3, TFH | 99 | |
| 5165 | G2553A | RNR2 | 17 | UCEC, Panc-AdenoCA, Prost-AdenoCA [31,33] |
| 5165 | G5753A | OL | 98 | HCC, Uterus-AdenoCA [33] |
| 5165 | G14453A | ND6/A74V | 98 | Missense, deleterious (1.0) d; pathogenic (0.81) e; Kidney-ChRCC, Ovary-AdenoCA, CNS-Medullo, HCC, Biliary-AdenoCA [33] |
| 5167 | T14325C | ND6/N117D | 94 | Missense, neutral (0.56) d; pathogenic (0.54) e; UCEC [31] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Young, M.J.; Sachidanandam, R.; Hales, D.B.; Brard, L.; Robinson, K.; Rahman, M.M.; Khadka, P.; Groesch, K.; Young, C.K.J. Identification of Somatic Mitochondrial DNA Mutations, Heteroplasmy, and Increased Levels of Catenanes in Tumor Specimens Obtained from Three Endometrial Cancer Patients. Life 2022, 12, 562. https://doi.org/10.3390/life12040562
Young MJ, Sachidanandam R, Hales DB, Brard L, Robinson K, Rahman MM, Khadka P, Groesch K, Young CKJ. Identification of Somatic Mitochondrial DNA Mutations, Heteroplasmy, and Increased Levels of Catenanes in Tumor Specimens Obtained from Three Endometrial Cancer Patients. Life. 2022; 12(4):562. https://doi.org/10.3390/life12040562
Chicago/Turabian StyleYoung, Matthew J., Ravi Sachidanandam, Dale B. Hales, Laurent Brard, Kathy Robinson, Md. Mostafijur Rahman, Pabitra Khadka, Kathleen Groesch, and Carolyn K. J. Young. 2022. "Identification of Somatic Mitochondrial DNA Mutations, Heteroplasmy, and Increased Levels of Catenanes in Tumor Specimens Obtained from Three Endometrial Cancer Patients" Life 12, no. 4: 562. https://doi.org/10.3390/life12040562