
Amphiphilic Cell-Penetrating Peptides Containing Natural and Unnatural Amino Acids as Drug Delivery Agents

 ,
,  , and
, and
Abstract
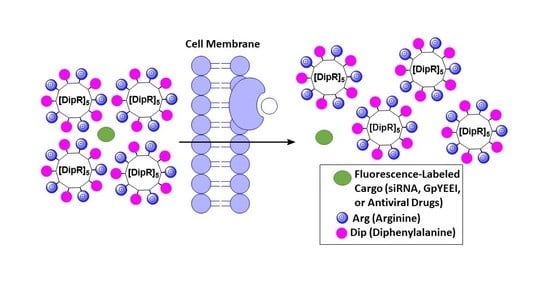
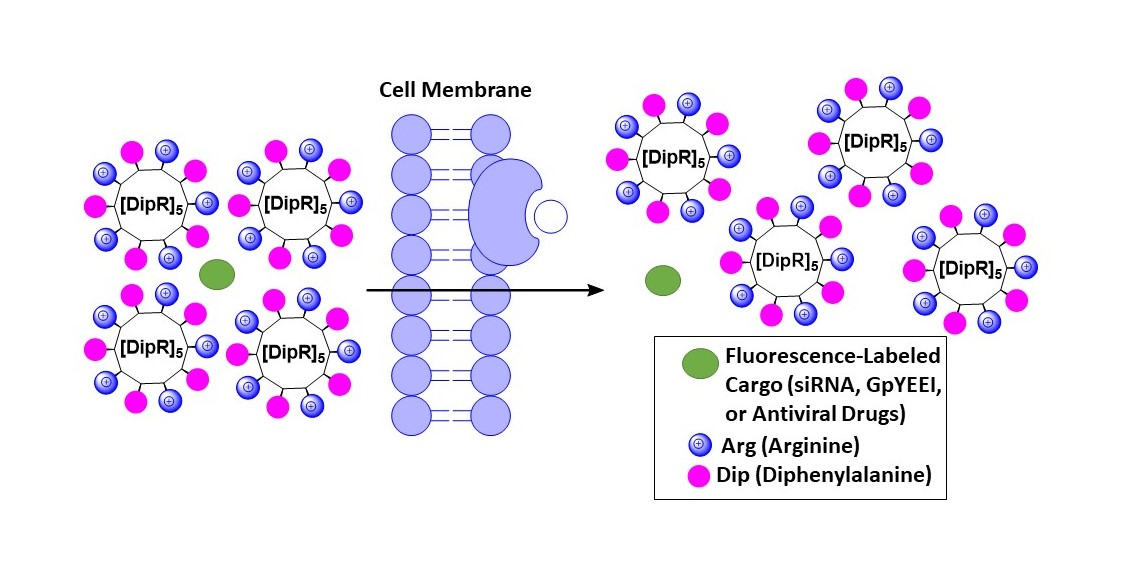
:
{kind=link}
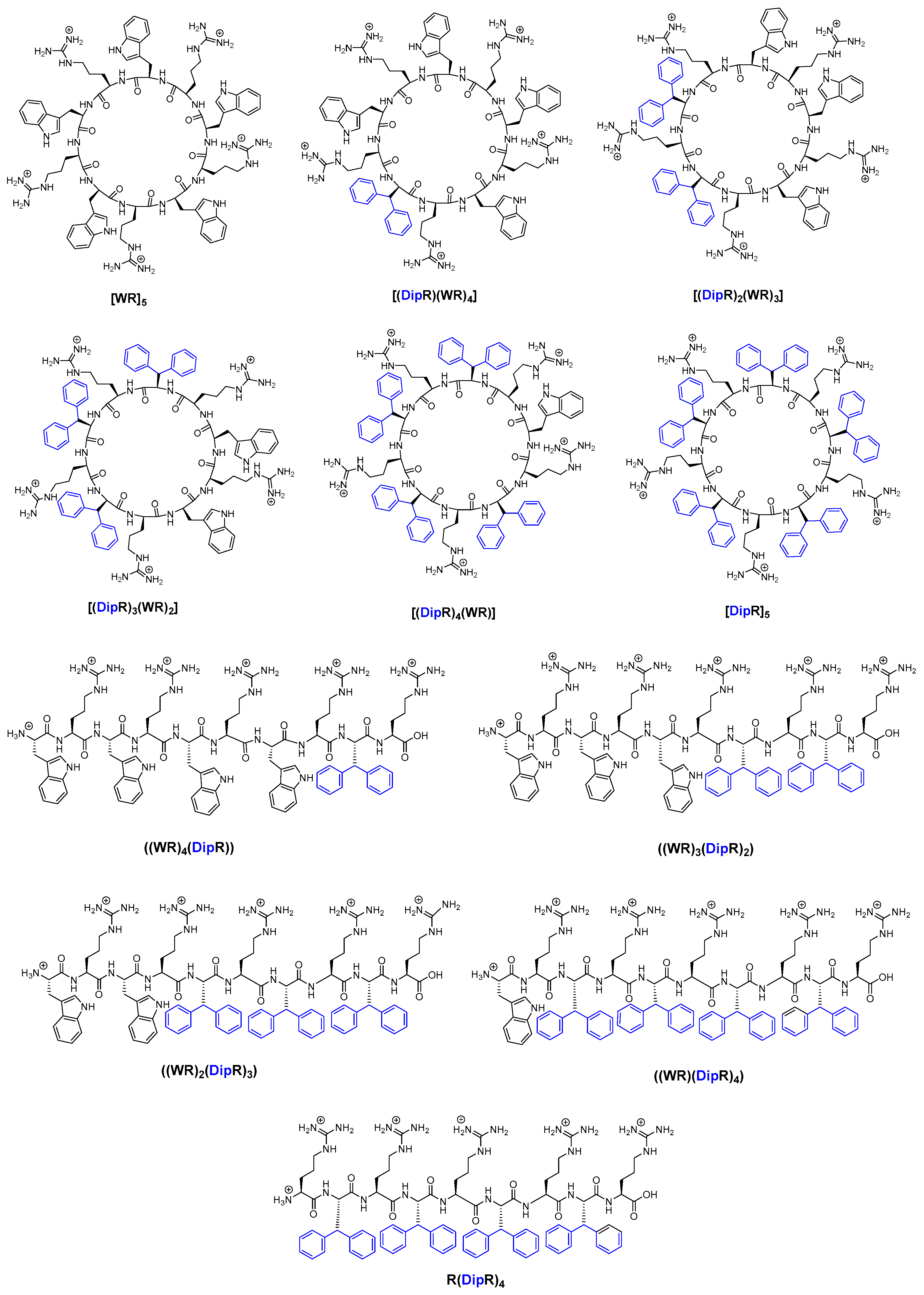{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
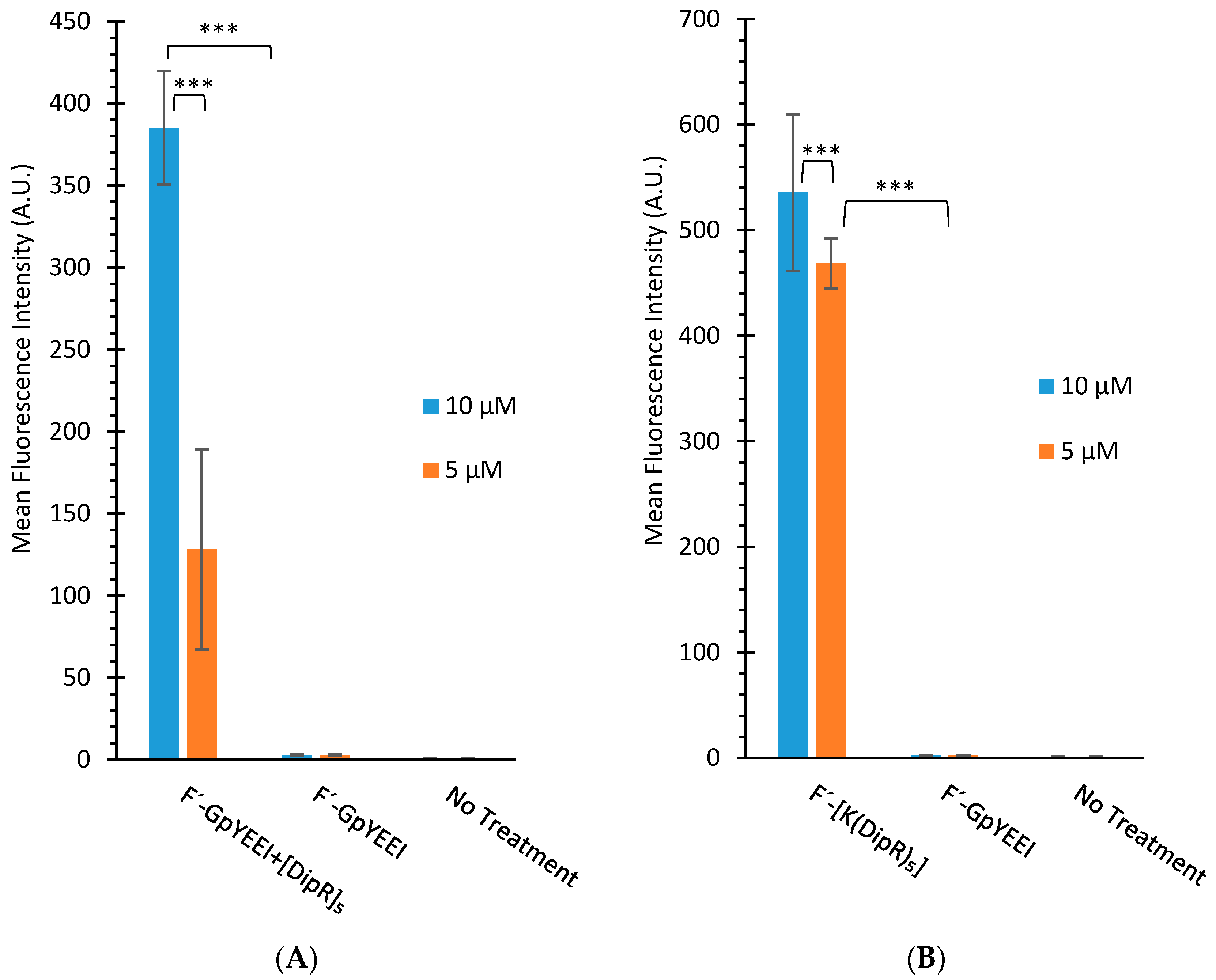{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. General
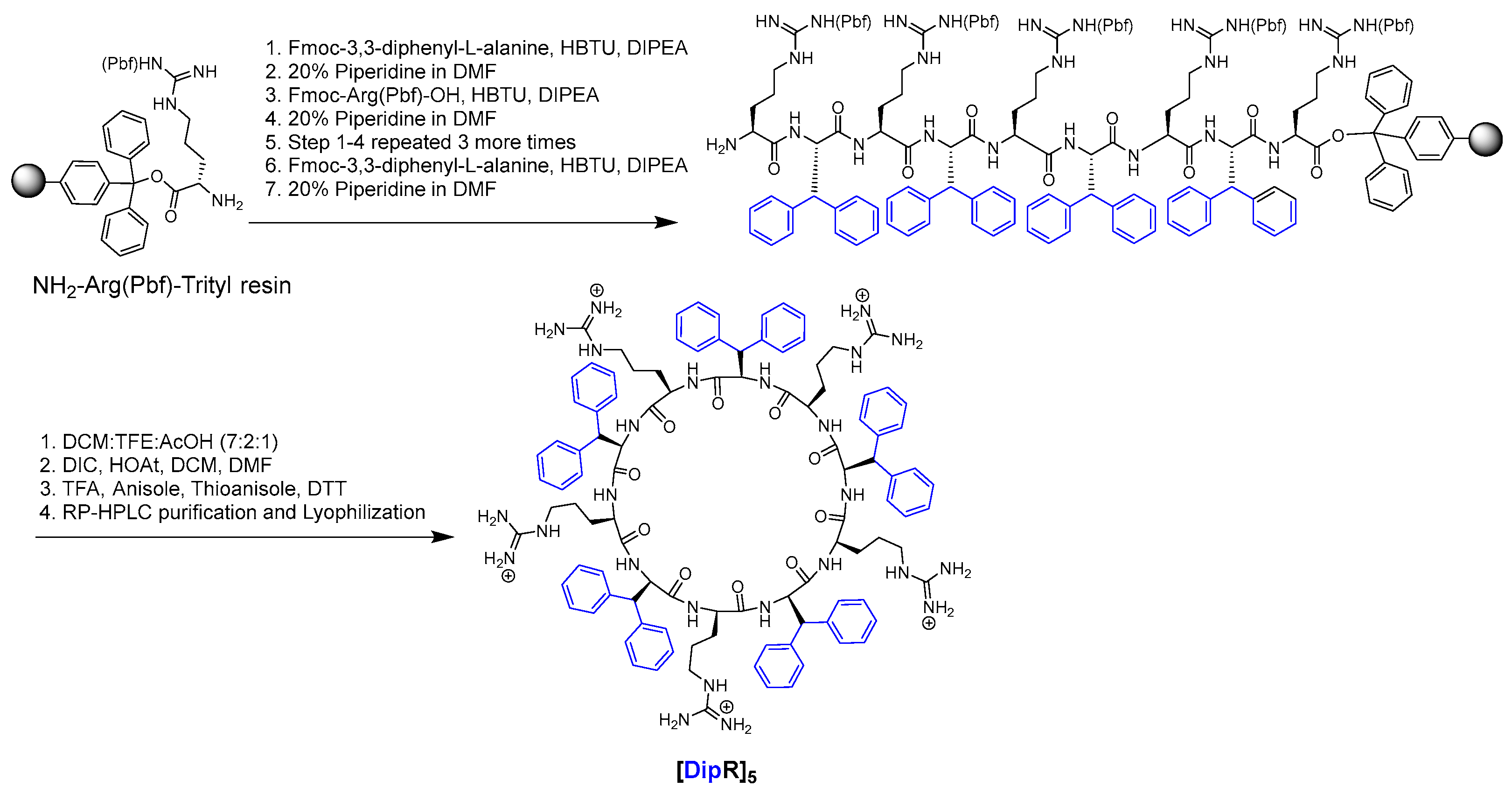
2.2. Synthesis of Peptides
2.3. In Vitro Cytotoxicity Assays
2.4. Cellular Uptake Studies
2.5. Mechanism of Cellular Uptake of Cargo
2.6. Circular Dichroism
2.7. Transmission Electron Microscopy
2.8. Dynamic Light Scattering
2.9. Confocal Microscopy
2.10. Plasma Stability Study
2.11. Data Analysis
3. Results and Discussion
3.1. Chemistry
3.2. Biological Activities
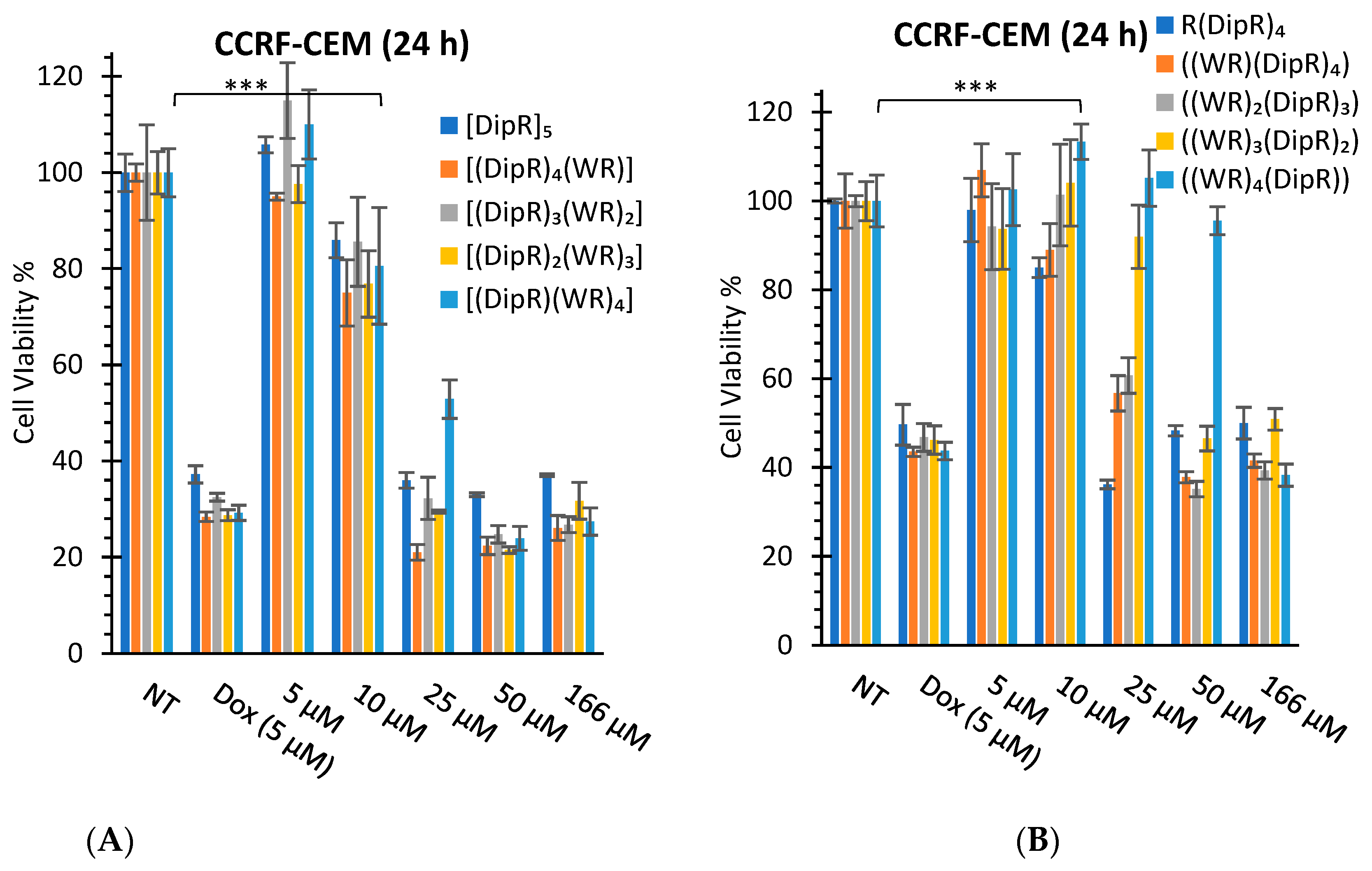
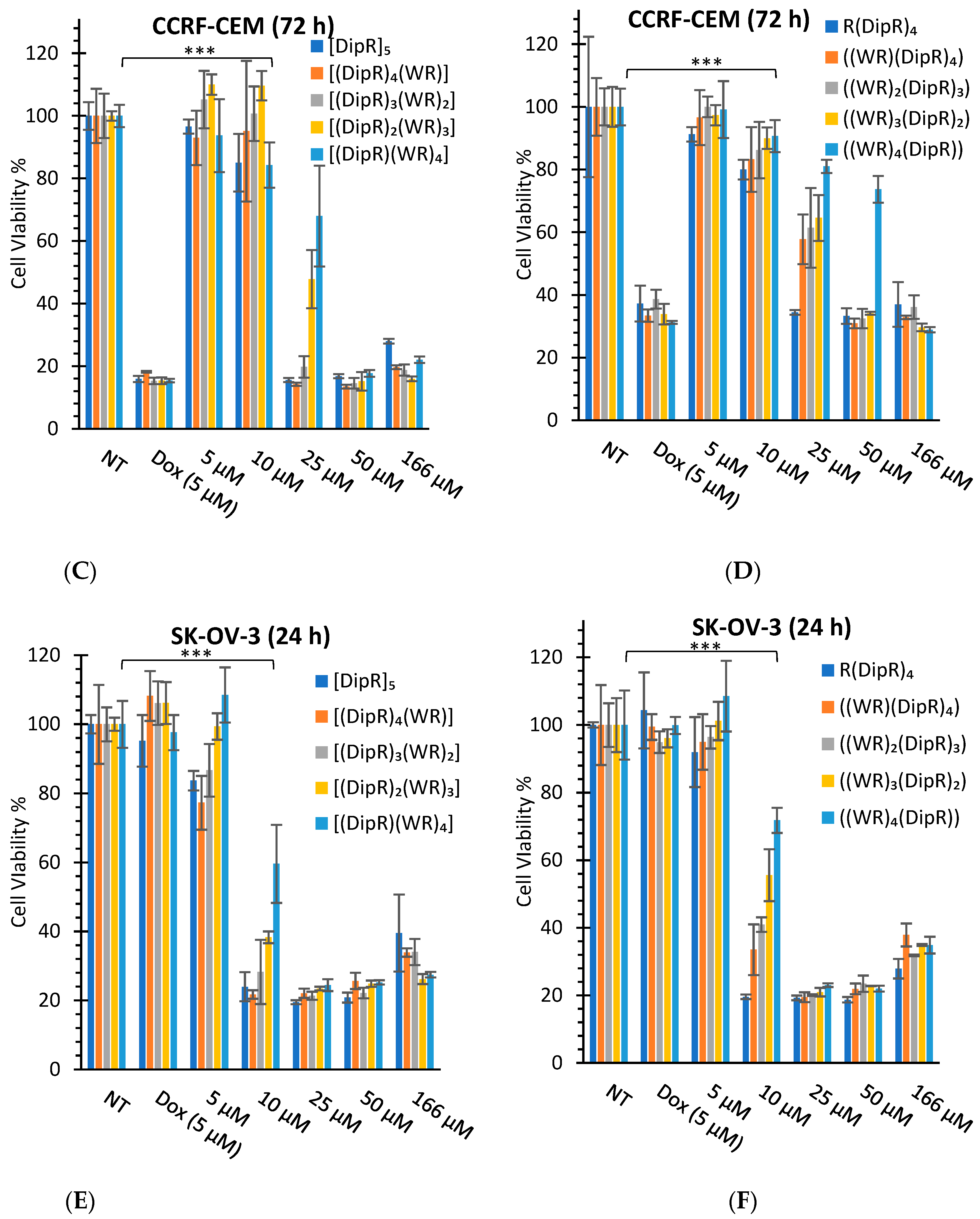
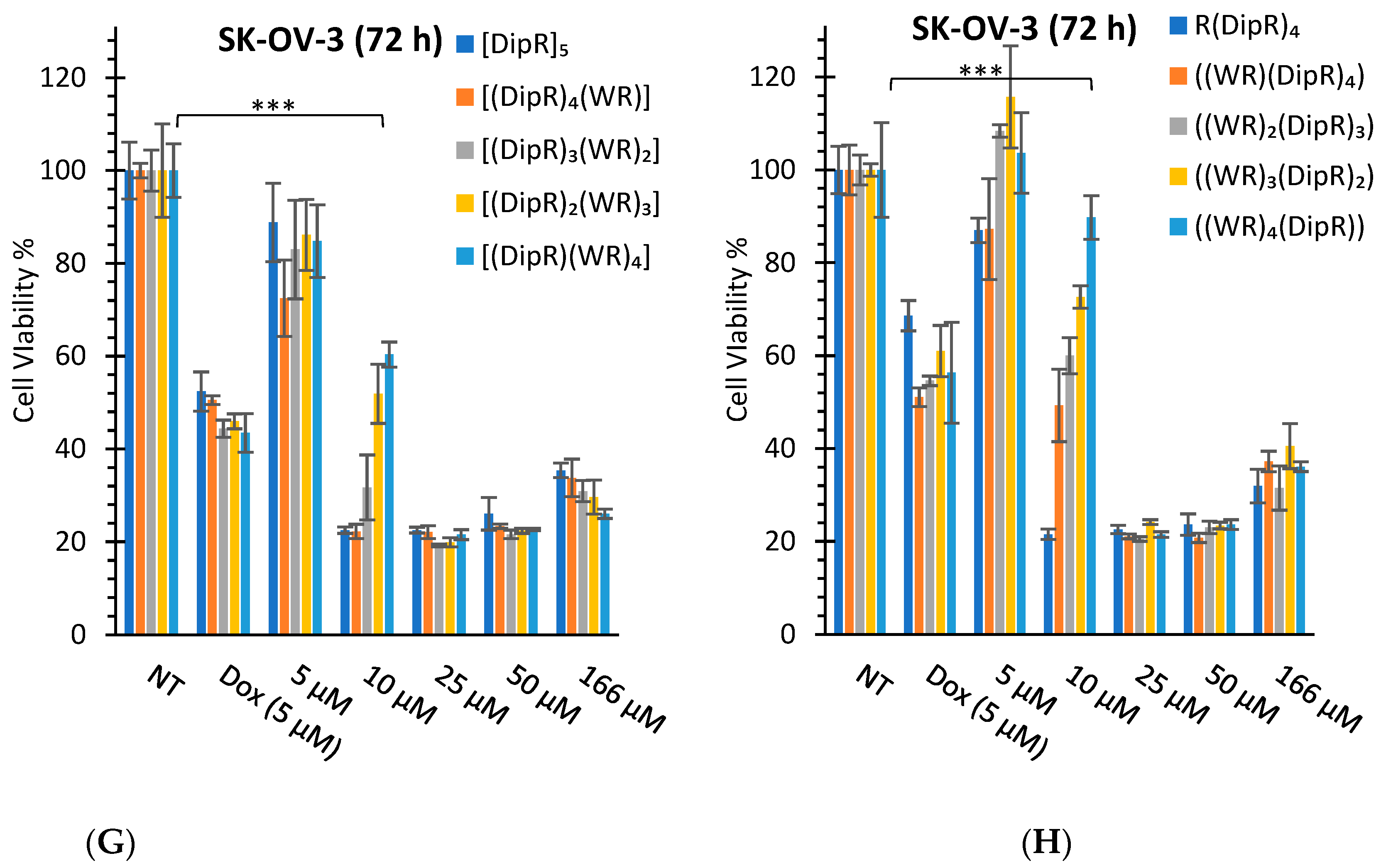
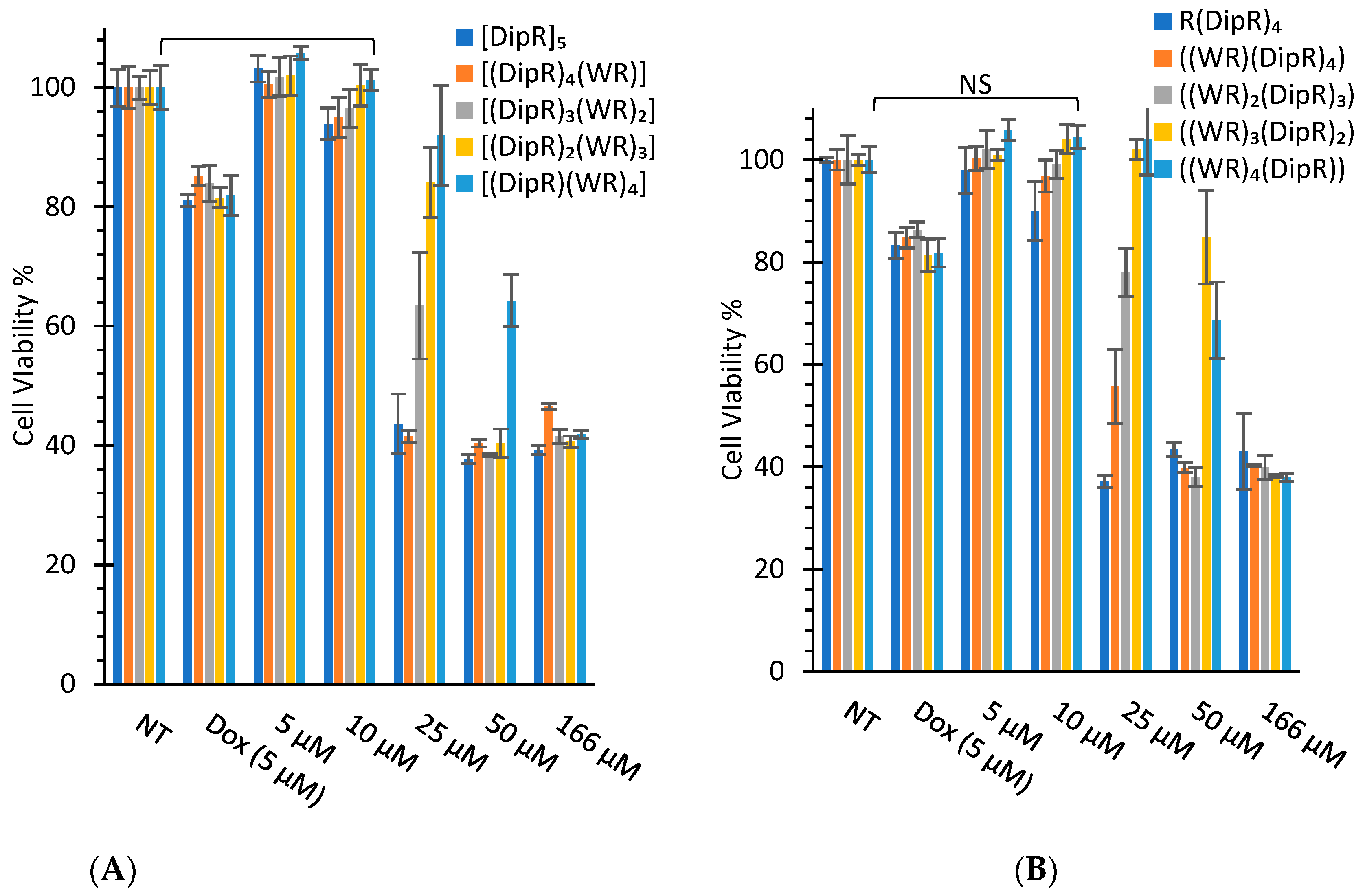
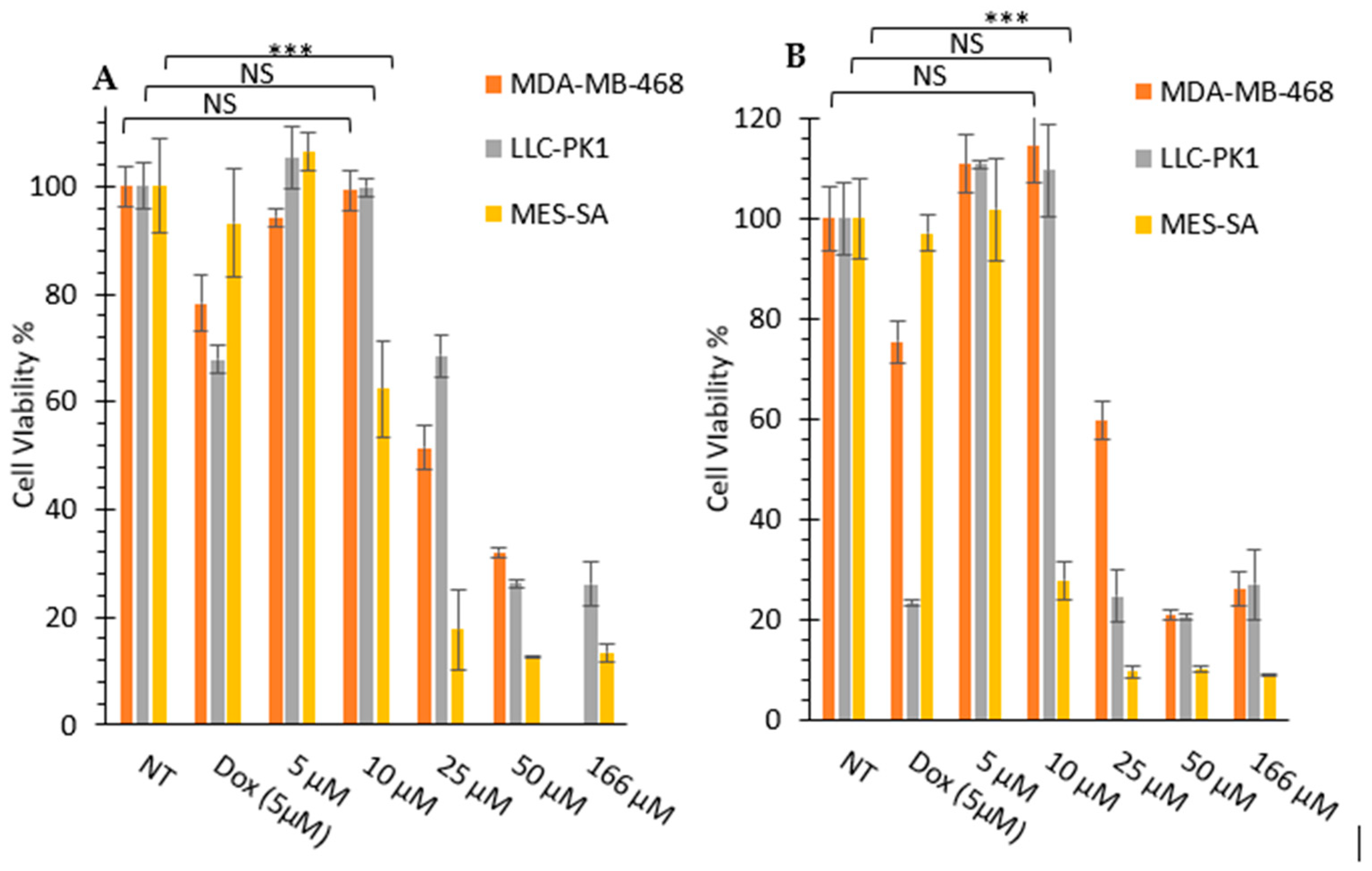
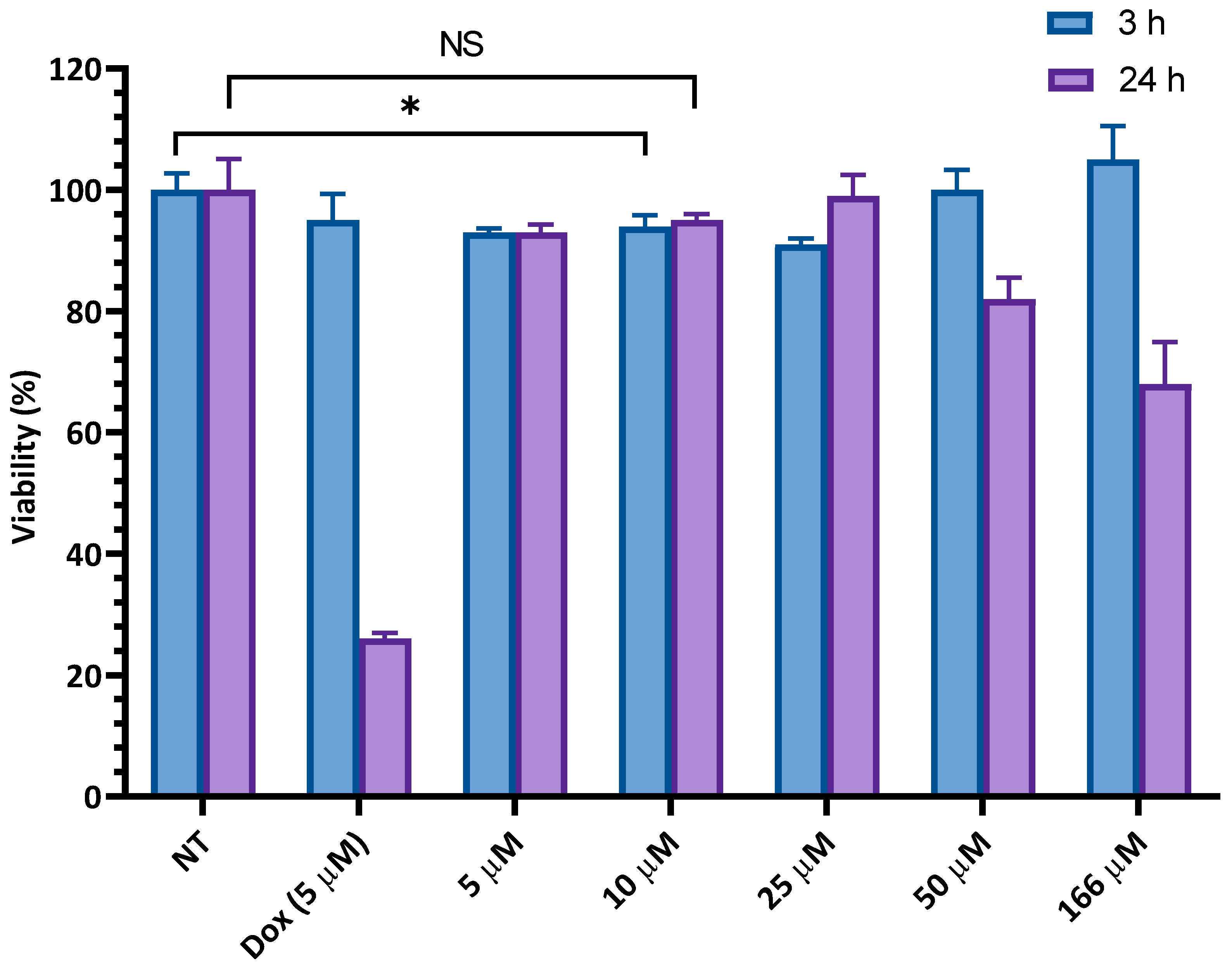
3.2.1. In Vitro Cytotoxicity Assays
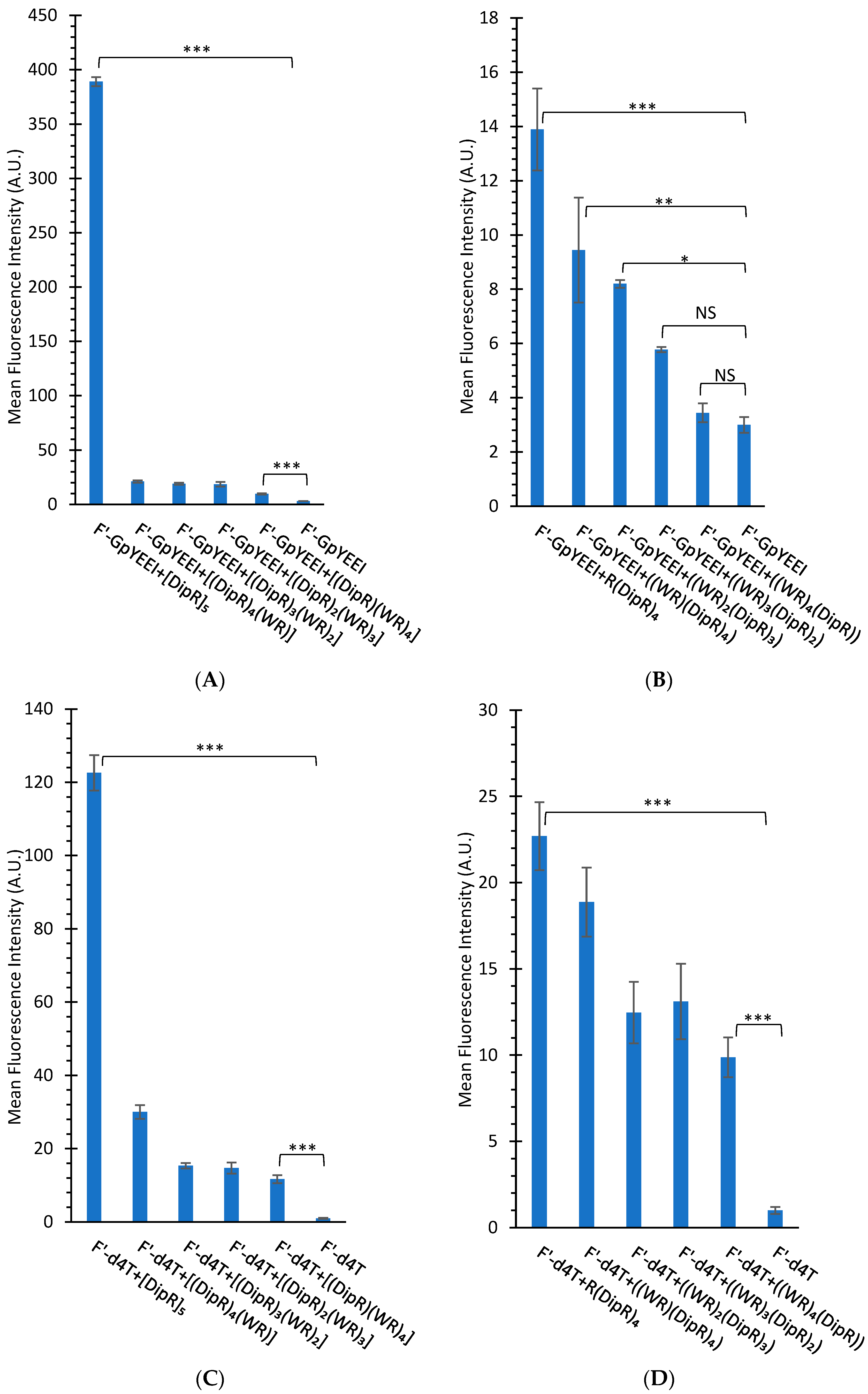
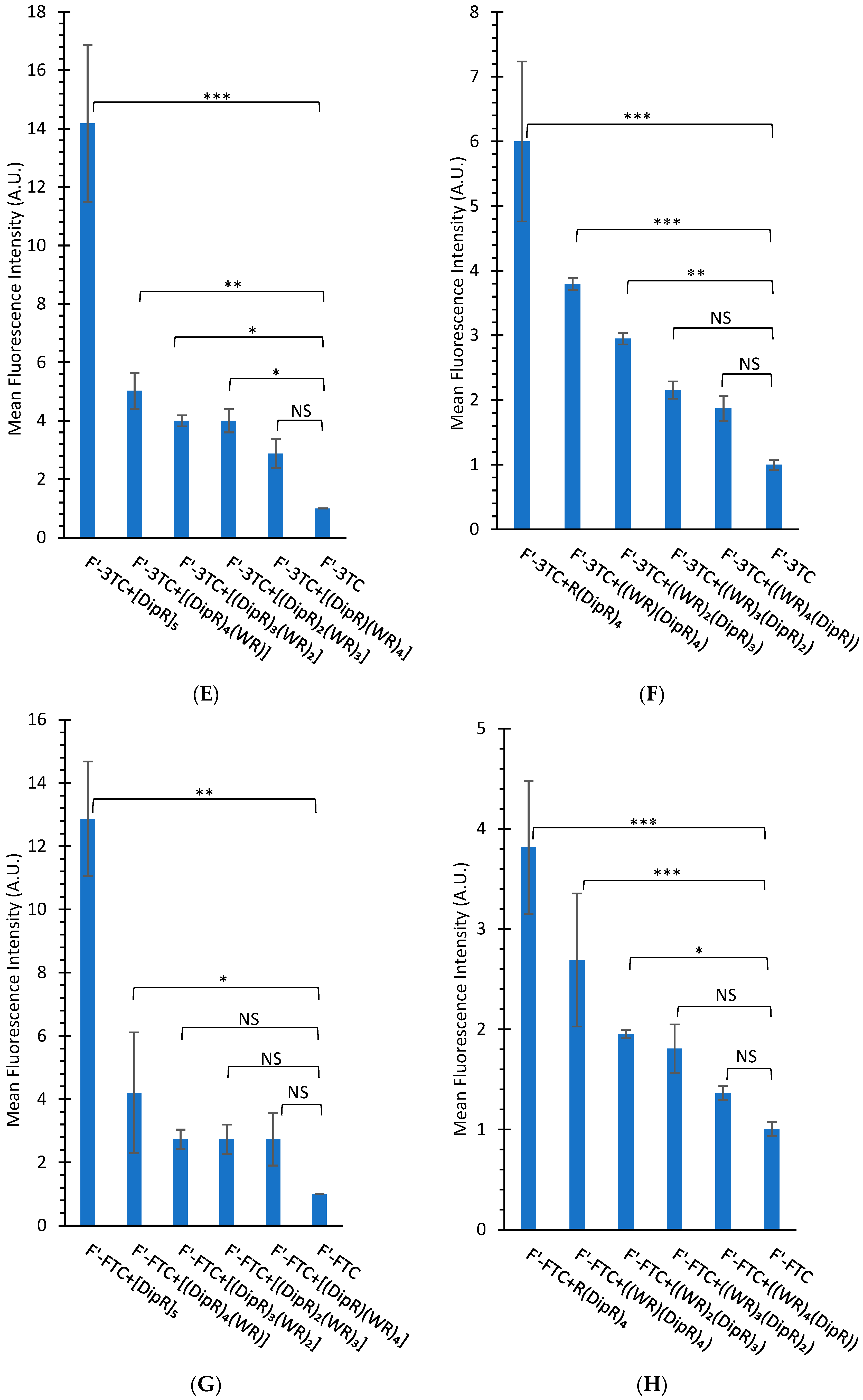
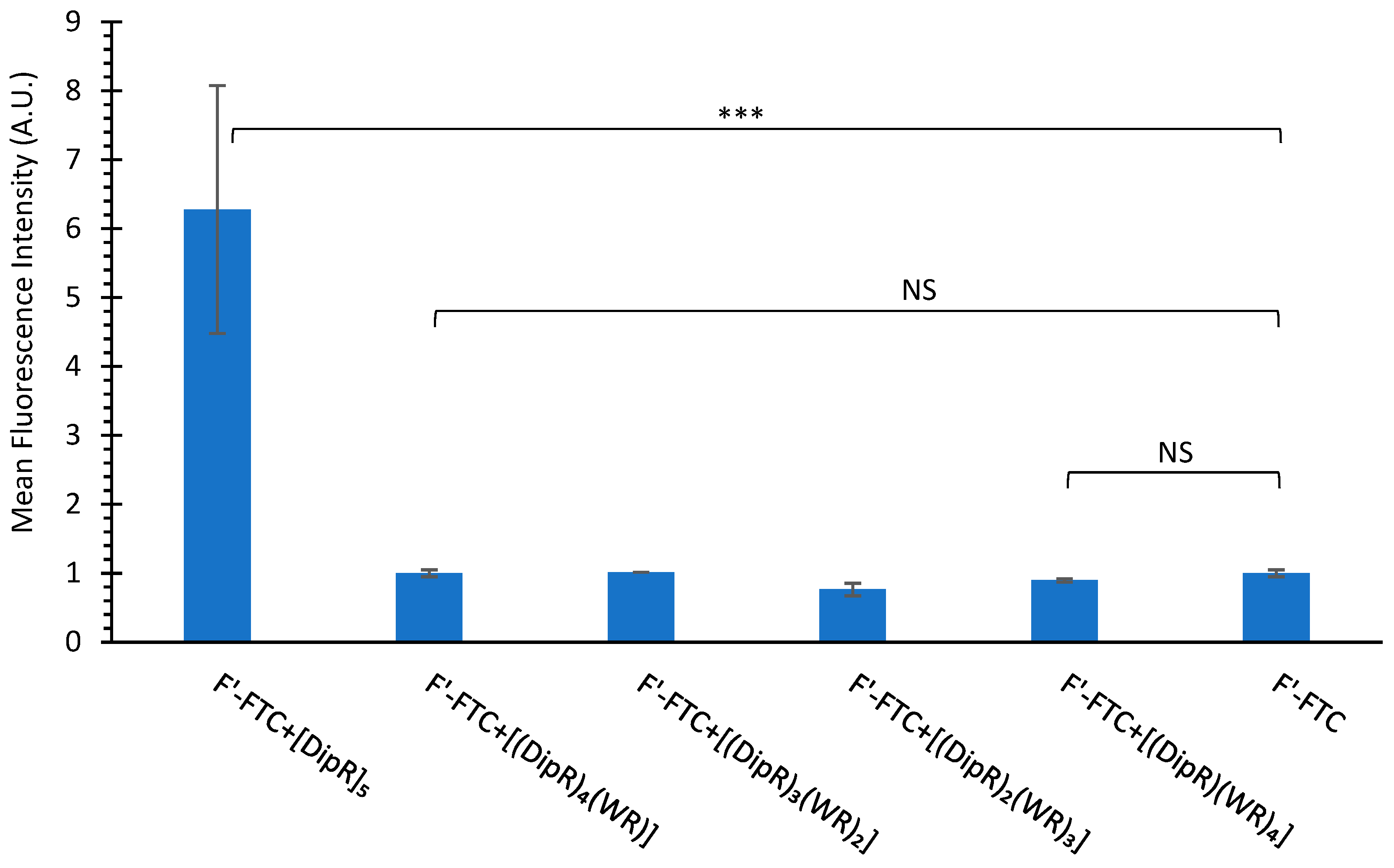
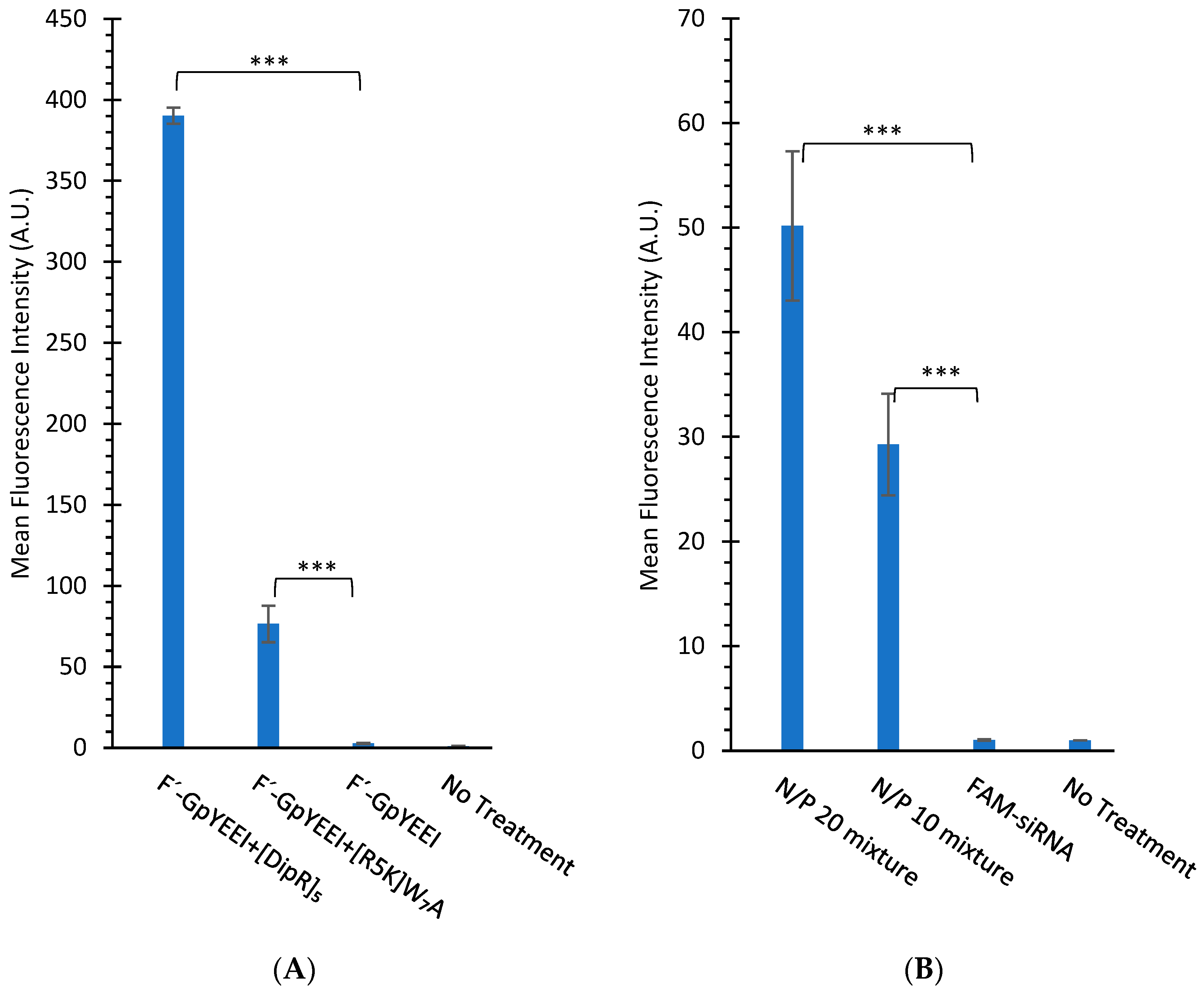
3.2.2. Cellular Uptake Studies
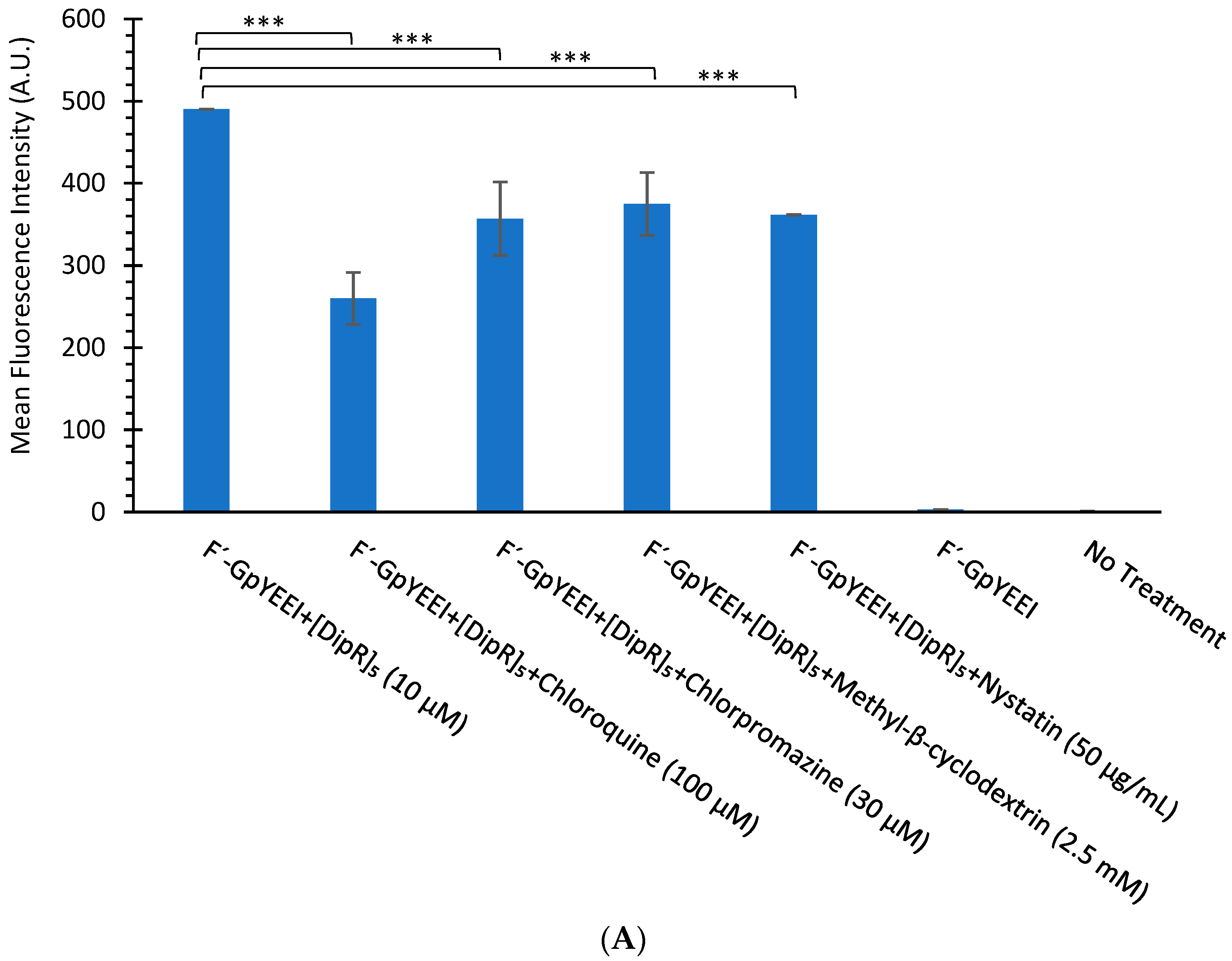
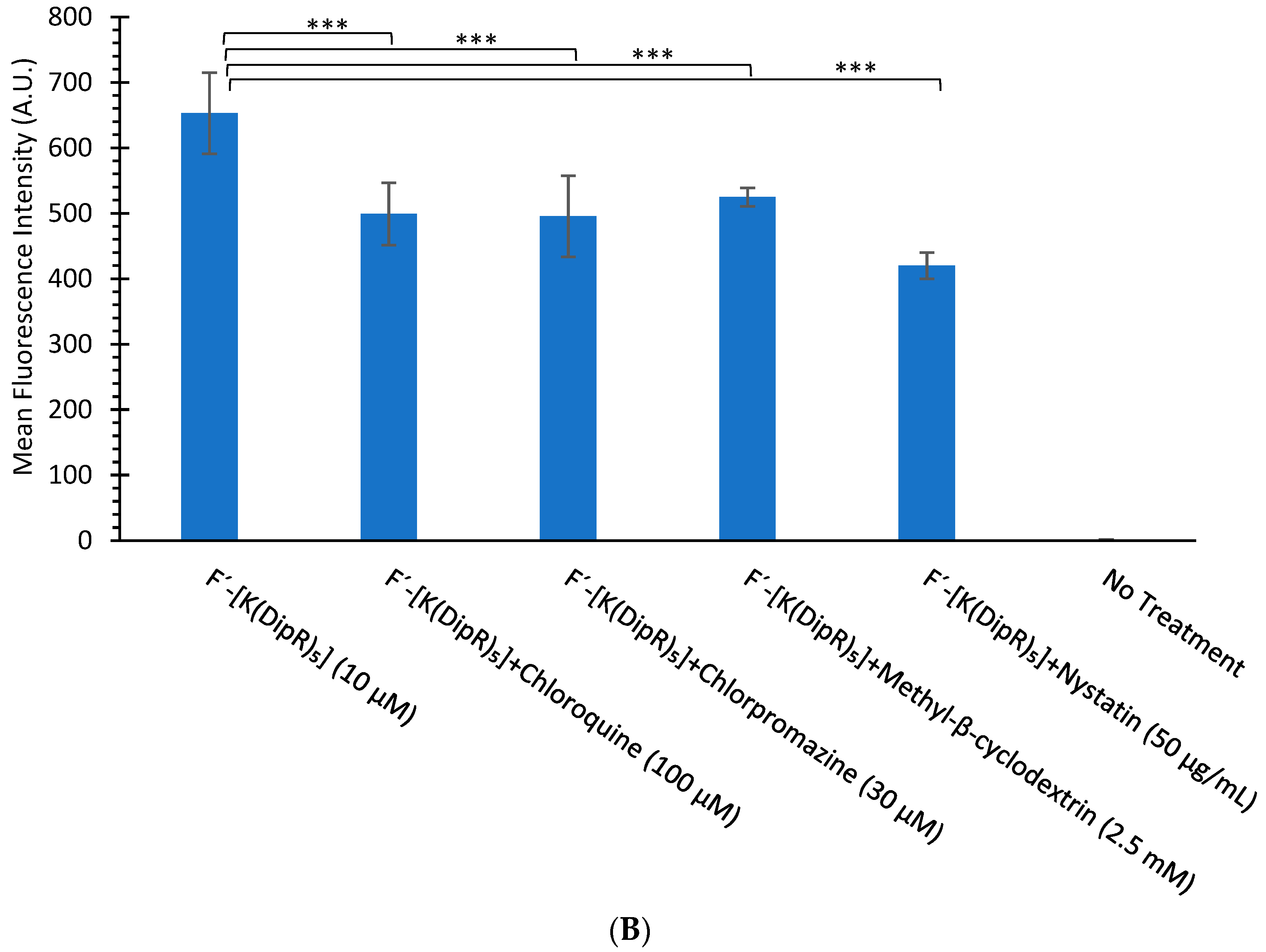
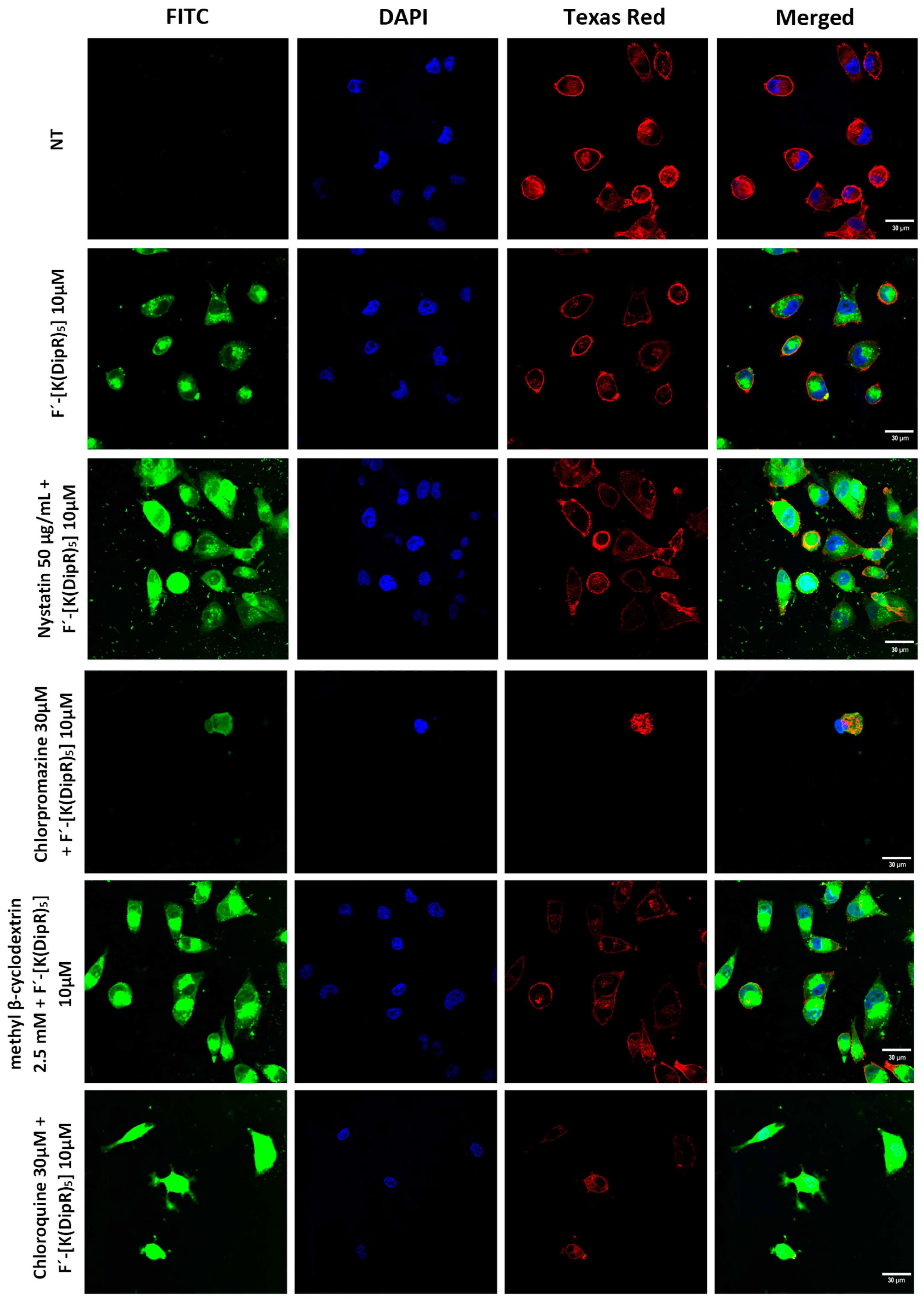
3.2.3. Mechanism of Cellular Uptake of Cargo
3.2.4. Circular Dichroism
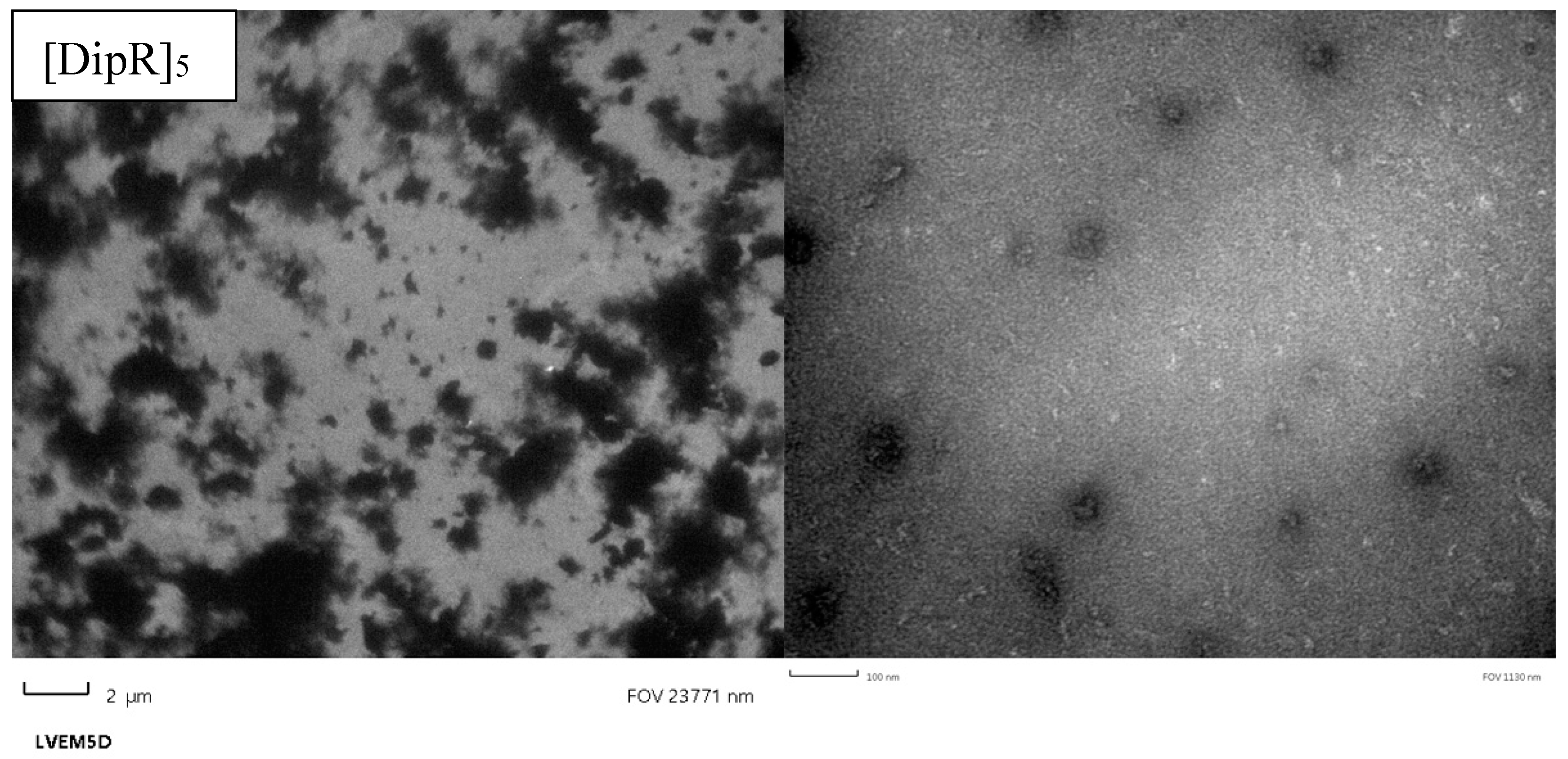
3.2.5. Transmission Electron Microscopy
3.2.6. Dynamic Light Scattering
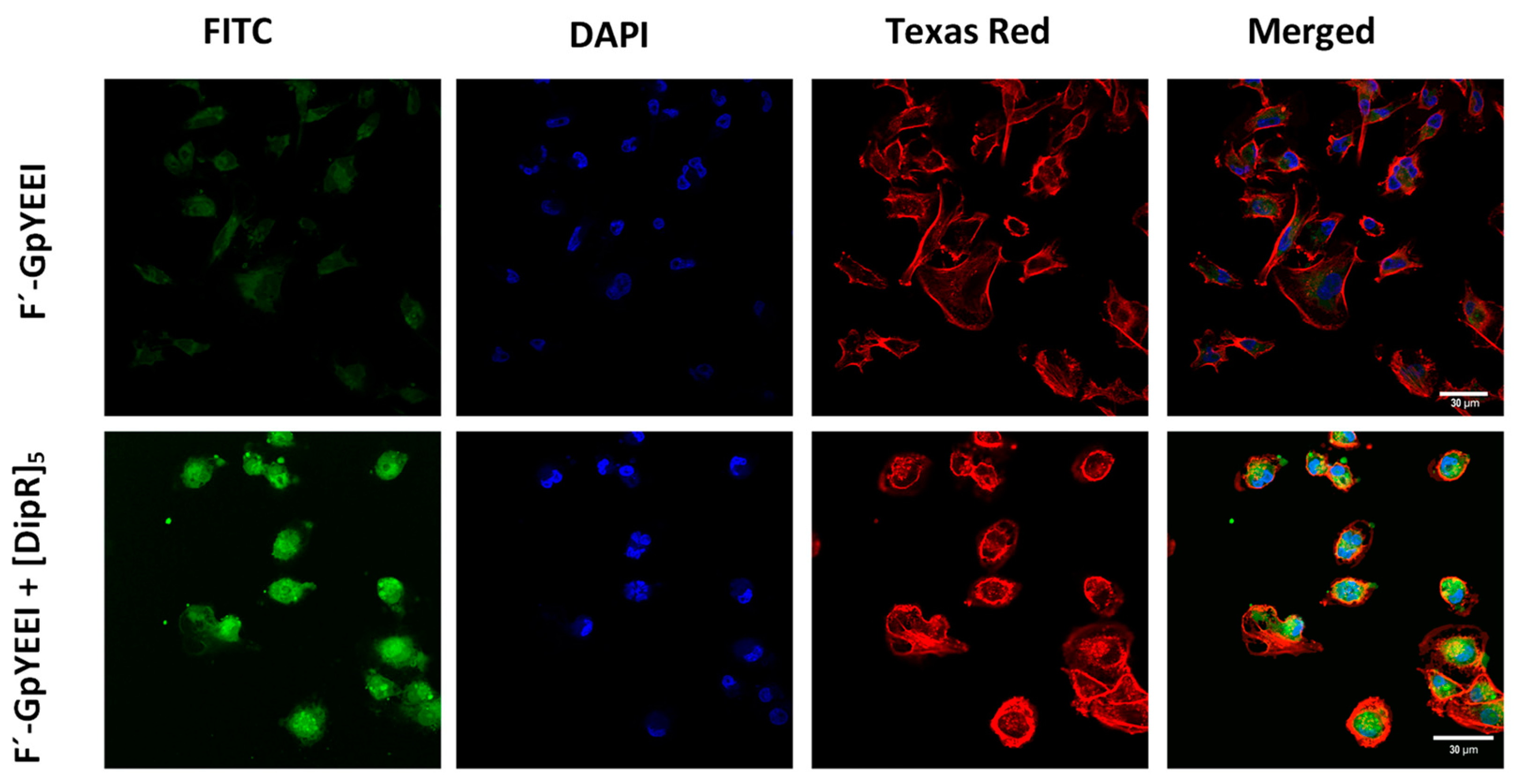
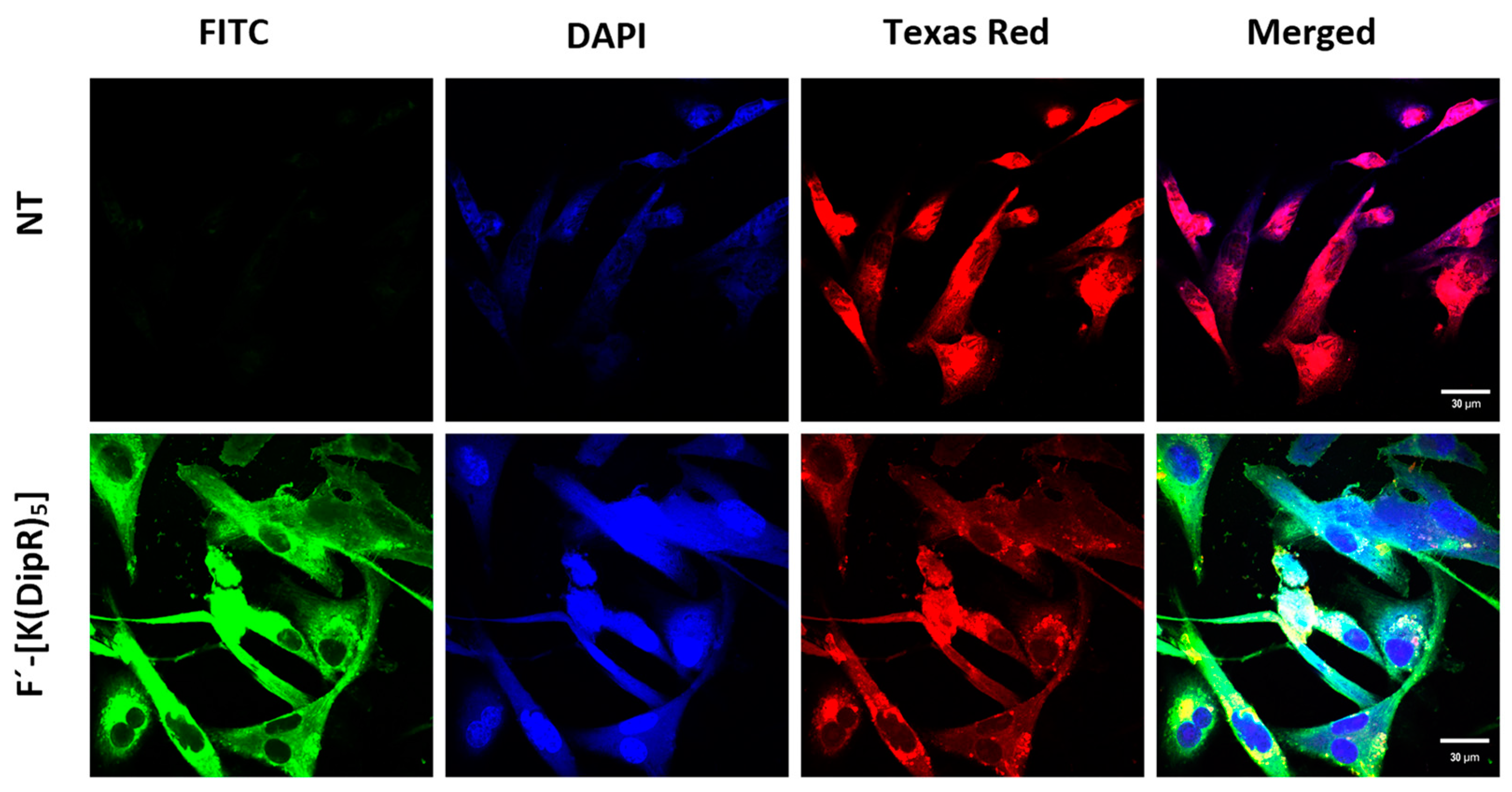
3.2.7. Confocal Microscopy
3.2.8. Plasma Stability of [DipR]5
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Y.; Cheetham, A.G.; Angacian, G.; Su, H.; Xie, L.; Cui, H. Peptide-drug conjugates as effective prodrug strategies for targeted delivery. Adv. Drug Deliv. Rev. 2017, 110–111, 112–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dissanayake, S.; Denny, W.A.; Gamage, S.; Sarojini, V. Recent developments in anticancer drug delivery using cell penetrating and tumor targeting peptides. J. Control Release 2017, 250, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Wiradharma, N.; Tong, Y.W.; Yang, Y.Y. Self-assembled oligopeptide nanostructuRes. for co-delivery of drug and gene with synergistic therapeutic effect. Biomaterials 2009, 30, 3100–3109. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.P.; Melikov, K.; Vives, E.; Ramos, C.; Verbeure, B.; Gait, M.J.; Chernomordik, L.V.; Lebleu, B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 2003, 278, 585–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torchilin, V.P.; Rammohan, R.; Weissig, V.; Levchenko, T.S. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc. Natl. Acad. Sci. USA 2001, 98, 8786–8791. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.H.; Cai, Y.Y.; Du, J.K.; Li, L.; Li, Q.; Yang, H.K.; Lin, J.T. TAT-conjugated chitosan cationic micelle for nuclear-targeted drug and gene co-delivery. Colloids Surf. B Biointerfaces 2018, 162, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Konishi, Y.; Ueda, M.; Saji, H.; Futaki, S. Accumulation of arginine-rich cell-penetrating peptides in tumors and the potential for anticancer drug delivery in vivo. J. Control Release 2012, 159, 181–188. [Google Scholar] [CrossRef]
- Zai, W.; Chen, W.; Wu, Z.; Jin, X.; Fan, J.; Zhang, X.; Luan, J.; Tang, S.; Mei, X.; Hao, Q.; et al. Targeted interleukin-22 gene delivery in the liver by polymetformin and penetratin-based hybrid nanoparticles to treat nonalcoholic fatty liver disease. ACS Appl. Mater. Interfaces 2019, 11, 4842–4857. [Google Scholar] [CrossRef]
- Wang, W.; Suga, T.; Hagimori, M.; Kuroda, N.; Fuchigami, Y.; Kawakami, S. Investigation of intracellular delivery of NuBCP-9 by conjugation with oligoarginines peptides in MDA-MB-231 cells. Biol. Pharm. Bull. 2018, 41, 1448–1455. [Google Scholar] [CrossRef]
- Thorén, P.E.; Persson, D.; Isakson, P.; Goksör, M.; Onfelt, A.; Nordén, B. Uptake of analogs of penetratin, Tat(48-60) and oligoarginine in live cells. BioChem. Biophys. Res. Commun. 2003, 307, 100–107. [Google Scholar] [CrossRef]
- Komin, A.; Russell, L.M.; Hristova, K.A.; Searson, P.C. Peptide-based strategies for enhanced cell uptake, transcellular transport, and circulation: Mechanisms and challenges. Adv. Drug Deliv. Rev. 2017, 110, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Choi, J.J.; Choi, Y.J.; Hennig, B.; Toborek, M. HIV-1 Tat-induced cerebrovascular toxicity is enhanced in mice with amyloid deposits. Neurobiol. Aging 2012, 33, 1579–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, G.H.; Mazzola, E.; Opoku-Nsiah, K.; Lammert, M.A.; Godes, M.; Neuberg, D.S.; Walensky, L.D. Biophysical determinants for cellular uptake of hydrocarbon-stapled peptide helices. Nat. Chem. Biol. 2016, 12, 845–852. [Google Scholar] [CrossRef] [Green Version]
- Futaki, S.; Suzuki, T.; Ohashi, W.; Yagami, T.; Tanaka, S.; Ueda, K.; Sugiura, Y. Arginine-rich peptides—An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 2001, 276, 5836–5840. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.B.; Moon, K.S.; Lee, M. Recent advances in functional supramolecular nanostructuRes. assembled from bioactive building blocks. Chem. Soc. Rev. 2009, 38, 925–934. [Google Scholar] [CrossRef]
- Walrant, A.; Cardon, S.; Burlina, F.; Sagan, S. Membrane crossing and membranotropic activity of cell-penetrating peptides: Dangerous liaisons? Acc. Chem. Res. 2017, 50, 2968–2975. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Aisenbrey, C.; Harmouche, N.; Raya, J.; Bertani, P.; Voievoda, N.; Süss, R.; Bechinger, B. pH-dependent membrane interactions of the histidine-rich cell-penetrating LAH4-L1. Biophys. J. 2017, 113, 1290–1300. [Google Scholar] [CrossRef] [Green Version]
- Via, M.A.; Klug, J.; Wilke, N.; Mayorga, L.S.; Del Pópolo, M.G. The interfacial electrostatic potential modulates the insertion of cell-penetrating peptides into lipid bilayers. Phys. Chem. Chem. Phys. 2018, 20, 5180–5189. [Google Scholar] [CrossRef]
- Esbjörner, E.K.; Lincoln, P.; Nordén, B. Counterion-mediated membrane penetration: Cationic cell-penetrating peptides overcome Born energy barrier by ion-pairing with phospholipids. Biochim. Biophys. Acta 2007, 1768, 1550–1558. [Google Scholar] [CrossRef] [Green Version]
- Ye, G.; Gupta, A.; DeLuca, R.; Parang, K.; Bothun, G.D. Bilayer disruption and liposome restructuring by a homologous series of small Arg-rich synthetic peptides. Colloids Surf. B Biointerfaces 2010, 76, 76–81. [Google Scholar] [CrossRef]
- Ye, G.; Nam, N.H.; Kumar, A.; Saleh, A.; Shenoy, D.B.; Amiji, M.M.; Lin, X.; Sun, G.; Parang, K. Synthesis and evaluation of tripodal peptide analogues for cellular delivery of phosphopeptides. J. Med. Chem. 2007, 50, 3604–3617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaroche, D.; Aussedat, B.; Aubry, S.; Chassaing, G.; Burlina, F.; Clodic, G.; Bolbach, G.; Lavielle, S.; Sagan, S. Tracking a new cell-penetrating (W/R) nonapeptide, through an enzyme-stable mass spectrometry reporter tag. Anal. Chem. 2007, 79, 1932–1938. [Google Scholar] [CrossRef] [PubMed]
- Thorén, P.E.; Persson, D.; Esbjörner, E.K.; Goksör, M.; Lincoln, P.; Nordén, B. Membrane binding and translocation of cell-penetrating peptides. Biochemistry 2004, 43, 3471–3489. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Kauffman, W.B.; Fuselier, T.; Naveen, S.K.; Voss, T.G.; Hristova, K.; Wimley, W.C. Direct cytosolic delivery of polar cargo to cells by spontaneous membrane-translocating peptides. J. Biol. Chem. 2013, 288, 29974–29986. [Google Scholar] [CrossRef] [Green Version]
- Nasrolahi Shirazi, A.; Tiwari, R.; Chhikara, B.S.; Mandal, D.; Parang, K. Design and biological evaluation of cell-penetrating peptide-doxorubicin conjugates as prodrugs. Mol. Pharm. 2013, 10, 488–499. [Google Scholar] [CrossRef]
- Mandal, D.; Nasrolahi Shirazi, A.; Parang, K. Cell-penetrating homochiral cyclic peptides as nuclear-targeting molecular transporters. Angew. Chem. Int. Ed. Engl. 2011, 50, 9633–9637. [Google Scholar] [CrossRef]
- Nasrolahi Shirazi, A.; Tiwari, R.K.; Oh, D.; Banerjee, A.; Yadav, A.; Parang, K. Efficient delivery of cell impermeable phosphopeptides by a cyclic peptide amphiphile containing tryptophan and arginine. Mol. Pharm. 2013, 10, 2008–2020. [Google Scholar] [CrossRef] [Green Version]
- Shirazi, A.N.; Paquin, K.L.; Howlett, N.G.; Mandal, D.; Parang, K. Cyclic peptide-capped gold nanoparticles for enhanced siRNA delivery. Molecules 2014, 19, 13319–13331. [Google Scholar] [CrossRef]
- Nasrolahi Shirazi, A.; Mandal, D.; Tiwari, R.K.; Guo, L.; Lu, W.; Parang, K. Cyclic peptide-capped gold nanoparticles as drug delivery systems. Mol. Pharm. 2013, 10, 500–511. [Google Scholar] [CrossRef]
- Nasrolahi Shirazi, A.; Tiwari, R.K.; Oh, D.; Sullivan, B.; Kumar, A.; Beni, Y.A.; Parang, K. Cyclic peptide-selenium nanoparticles as drug transporters. Mol. Pharm. 2014, 11, 3631–3641. [Google Scholar] [CrossRef]
- Mandal, D.; Tiwari, R.K.; Shirazi, A.N.; Oh, D.; Ye, G.; Banerjee, A.; Yadav, A.; Parang, K. Self-assembled surfactant cyclic peptide nanostructures as stabilizing agents. Soft Matter. 2013, 9, 9465–9475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoghebi, K.; Aliabadi, H.M.; Tiwari, R.K.; Parang, K. [(WR)(8)WKβA]-doxorubicin conjugate: A delivery system to overcome multi-drug resistance against doxorubicin. Cells 2022, 11, 301. [Google Scholar] [CrossRef] [PubMed]
- Mozaffari, S.; Salehi, D.; Mahdipoor, P.; Beuttler, R.; Tiwari, R.; Aliabadi, H.M.; Parang, K. Design and application of hybrid cyclic-linear peptide-doxorubicin conjugates as a strategy to overcome doxorubicin resistance and toxicity. Eur. J. Med. Chem. 2021, 226, 113836. [Google Scholar] [CrossRef]
- Darwish, S.; Sadeghiani, N.; Fong, S.; Mozaffari, S.; Hamidi, P.; Withana, T.; Yang, S.; Tiwari, R.K.; Parang, K. Synthesis and antiproliferative activities of doxorubicin thiol conjugates and doxorubicin-SS-cyclic peptide. Eur. J. Med. Chem. 2019, 161, 594–606. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, N.S.; Shirazi, A.N.; Sajid, M.I.; Park, S.E.; Parang, K.; Tiwari, R.K. Synthesis and antiproliferative activities of conjugates of paclitaxel and camptothecin with a cyclic cell-penetrating peptide. Molecules 2019, 24, 1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirazi, A.N.; El-Sayed, N.S.; Tiwari, R.K.; Tavakoli, K.; Parang, K. Cyclic peptide containing hydrophobic and positively charged residues as a drug delivery system for curcumin. Curr. Drug Deliv. 2016, 13, 409–417. [Google Scholar] [CrossRef]
- Hanna, S.E.; Mozaffari, S.; Tiwari, R.K.; Parang, K. Comparative molecular transporter efficiency of cyclic peptides containing tryptophan and arginine residues. ACS Omega 2018, 3, 16281–16291. [Google Scholar] [CrossRef]
- Molbase. Product Information: Tryptophan and Diphenylalanine. 2020. Available online: https://www.molbase.com/ (accessed on 10 January 2022).
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-penetrating peptides: From basic research to clinics. Trends Pharm. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef]
- Kong, J.; Wang, Y.F.; Qi, W.; Su, R.X.; He, Z.M. Photo- and aromatic stacking-induced green emissive peptidyl nanoparticles for cell imaging and monitoring of nucleic acid delivery. ACS Appl. Mater. Interfaces 2019, 11, 15401–15410. [Google Scholar] [CrossRef]
- Kaur, G.; Shukla, A.; Sivakumar, S.; Verma, S. Soft structure formation and cancer cell transport mechanisms of a folic acid-dipeptide conjugate. J. Pept. Sci. 2015, 21, 248–255. [Google Scholar] [CrossRef]
- Gour, N.; Kedracki, D.; Safir, I.; Ngo, K.X.; Vebert-Nardin, C. Self-assembling DNA-peptide hybrids: Morphological consequences of oligonucleotide grafting to a pathogenic amyloid fibrils forming dipeptide. Chem. Commun. 2012, 48, 5440–5442. [Google Scholar] [CrossRef] [PubMed]
- Emtiazi, G.; Zohrabi, T.; Lee, L.Y.; Habibi, N.; Zarrabi, A. Covalent diphenylalanine peptide nanotube conjugated to folic acid/magnetic nanoparticles for anti-cancer drug delivery. J. Drug Deliv. Sci. Technol. 2017, 41, 90–98. [Google Scholar] [CrossRef]
- Choe, R.; Yun, S.I. Fmoc-diphenylalanine-based hydrogels as a potential carrier for drug delivery. e-Polymers 2020, 20, 458–468. [Google Scholar] [CrossRef]
- Zhang, H.; Fei, J.B.; Yan, X.H.; Wang, A.H.; Li, J.B. Enzyme-responsive release of doxorubicin from monodisperse dipeptide-based nanocarriers for highly efficient cancer treatment in vitro. Adv. Funct. Mater. 2015, 25, 1193–1204. [Google Scholar] [CrossRef]
- Tesauro, D.; Accardo, A.; Diaferia, C.; Milano, V.; Guillon, J.; Ronga, L.; Rossi, F. Peptide-based drug-delivery systems in biotechnological applications: Recent advances and perspectives. Molecules 2019, 24, 351. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhao, J.; Wang, A.H.; Li, Q.; Cui, W. Supramolecular assembly of protein-based nanoparticles based on tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) for cancer therapy. Colloids Surf. A-Physicochem. Eng. Asp. 2020, 590, 124486. [Google Scholar] [CrossRef]
- Guan, S.W.; Yu, X.X.; Li, J.Y.; Xu, H.; Han, W.Z.; Shi, G.N.; Xu, J.; Wang, L.P. Delivery of survivin siRNA using cationic diphenylalanine vesicles. Chem. Res. Chin. Univ. 2019, 35, 434–439. [Google Scholar] [CrossRef]
- Ye, G.; Schuler, A.D.; Ahmadibeni, Y.; Morgan, J.R.; Faruqui, A.; Huang, K.; Sun, G.; Zebala, J.A.; Parang, K. Synthesis and evaluation of phosphopeptides containing iminodiacetate groups as binding ligands of the Src SH2 domain. Bioorg Chem. 2009, 37, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, H.K.; Chhikara, B.S.; Bhavaraju, S.; Mandal, D.; Doncel, G.F.; Parang, K. Emtricitabine prodrugs with improved anti-HIV activity and cellular uptake. Mol. Pharm. 2013, 10, 467–476. [Google Scholar] [CrossRef]
- Agarwal, H.K.; Chhikara, B.S.; Hanley, M.J.; Ye, G.F.; Doncel, G.F.; Parang, K. Synthesis and biological evaluation of fatty acyl ester derivatives of (-)-2’,3’-dideoxy-3‘-thiacytidine. J. Med. Chem. 2012, 55, 4861–4871. [Google Scholar] [CrossRef]
- Agarwal, H.K.; Loethan, K.; Mandal, D.; Doncel, G.F.; Parang, K. Synthesis and biological evaluation of fatty acyl ester derivatives of 2’,3‘-didehydro-2’,3’-dideoxythymidine. Bioorg. Med. Chem. Lett. 2011, 21, 1917–1921. [Google Scholar] [CrossRef] [PubMed]
- Mozaffari, S.; Bousoik, E.; Amirrad, F.; Lamboy, R.; Coyle, M.; Hall, R.; Alasmari, A.; Mahdipoor, P.; Parang, K.; Aliabadi, H.M. Amphiphilic peptides for efficient siRNA delivery. Polymers 2019, 11, 703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanan, G.; Merrill, D.; An, R.; Nolte, D.D.; Turek, J.J. Intracellular doppler spectroscopy detects altered drug response in SKOV3 tumor spheroids with silenced or inhibited P-glycoprotein. Biochem. Biophys. Res. Commun. 2019, 514, 1154–1159. [Google Scholar] [CrossRef]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006, 1, 2876–2890. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.R.; Lo, S.Y.; Liu, C.C.; Chyan, C.L.; Huang, Y.W.; Aronstam, R.S.; Lee, H.J. Endocytic trafficking of nanoparticles delivered by cell-penetrating peptides comprised of nona-arginine and a penetration accelerating sequence. PLoS ONE 2013, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raymond, D.M.; Nilsson, B.L. Multicomponent peptide assemblies. Chem. Soc. Rev. 2018, 47, 3659–3720. [Google Scholar] [CrossRef]
- Kim, S.; Huang, J.; Lee, Y.; Dutta, S.; Yoo, H.Y.; Jung, Y.M.; Jho, Y.; Zeng, H.; Hwang, D.S. Complexation and coacervation of like-charged polyelectrolytes inspired by mussels. Proc. Natl. Acad. Sci. USA 2016, 113, E847–E853. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.H.; Rothberg, K.G.; Anderson, R.G. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J. Cell Biol. 1993, 123, 1107–1117. [Google Scholar] [CrossRef]




















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salehi, D.; Mozaffari, S.; Zoghebi, K.; Lohan, S.; Mandal, D.; Tiwari, R.K.; Parang, K. Amphiphilic Cell-Penetrating Peptides Containing Natural and Unnatural Amino Acids as Drug Delivery Agents. Cells 2022, 11, 1156. https://doi.org/10.3390/cells11071156
Salehi D, Mozaffari S, Zoghebi K, Lohan S, Mandal D, Tiwari RK, Parang K. Amphiphilic Cell-Penetrating Peptides Containing Natural and Unnatural Amino Acids as Drug Delivery Agents. Cells. 2022; 11(7):1156. https://doi.org/10.3390/cells11071156
Chicago/Turabian StyleSalehi, David, Saghar Mozaffari, Khalid Zoghebi, Sandeep Lohan, Dindyal Mandal, Rakesh K. Tiwari, and Keykavous Parang. 2022. "Amphiphilic Cell-Penetrating Peptides Containing Natural and Unnatural Amino Acids as Drug Delivery Agents" Cells 11, no. 7: 1156. https://doi.org/10.3390/cells11071156