
Evaluation of Somatic Mutations in Solid Metastatic Pan-Cancer Patients

 , , , , , , ,
, , , , , , ,  add
Show full author list
add
Show full author list
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Characteristics
2.2. Statistical Analysis
3. Results
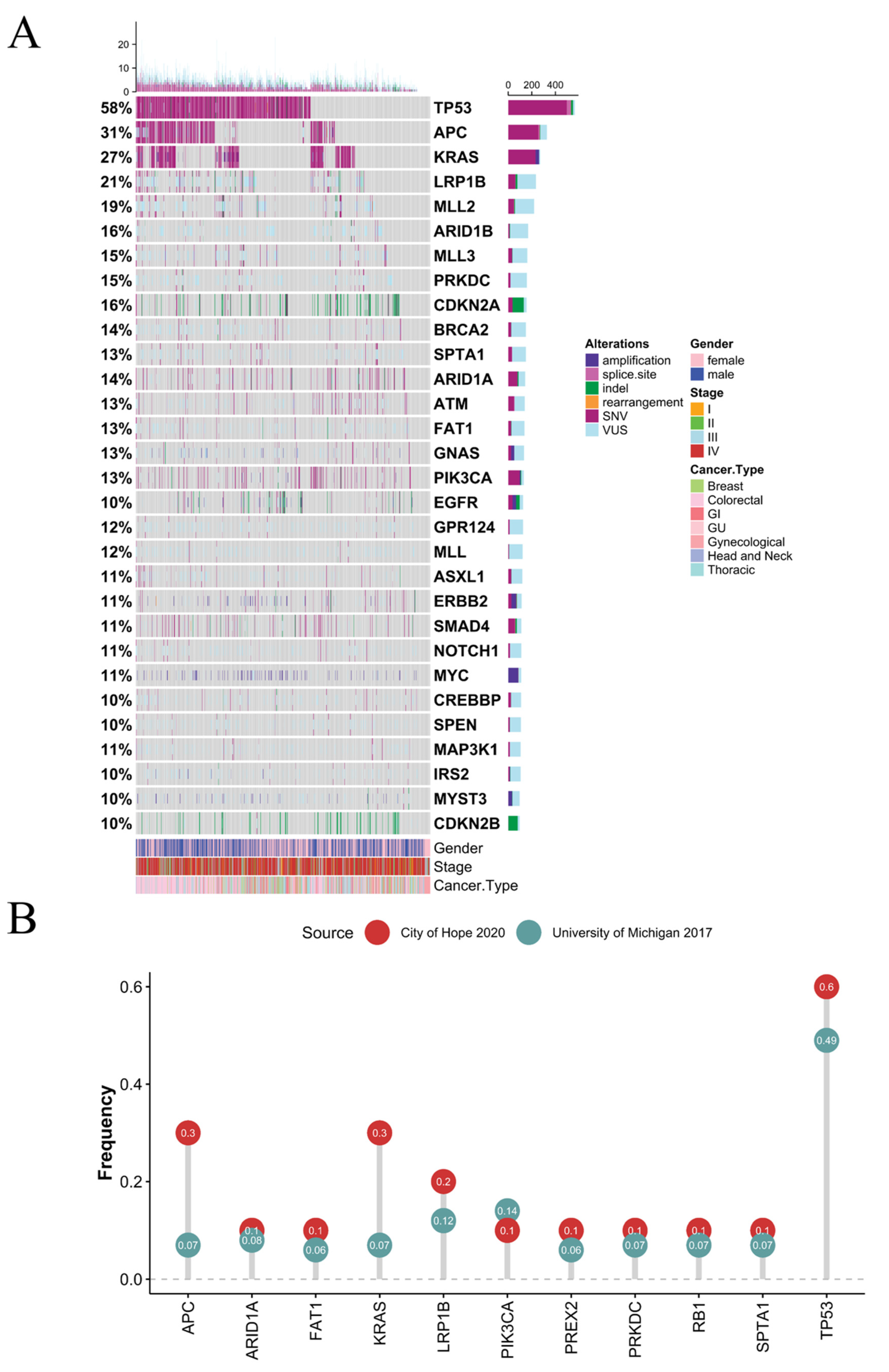
3.1. Clinical and Genomic Features
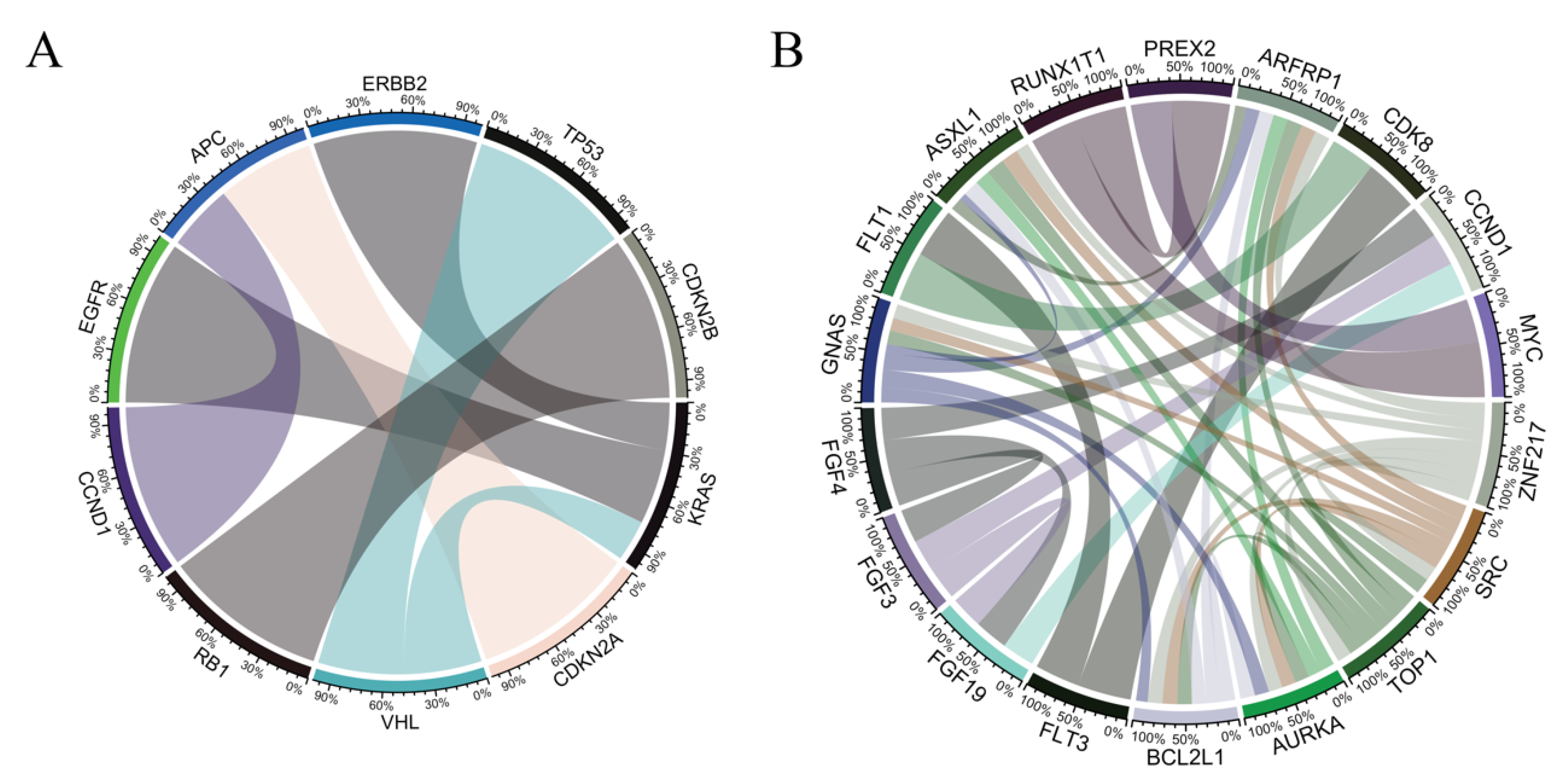
3.2. Co-Occurrence and Mutual-Exclusivity in Pan-Cancer Cohort
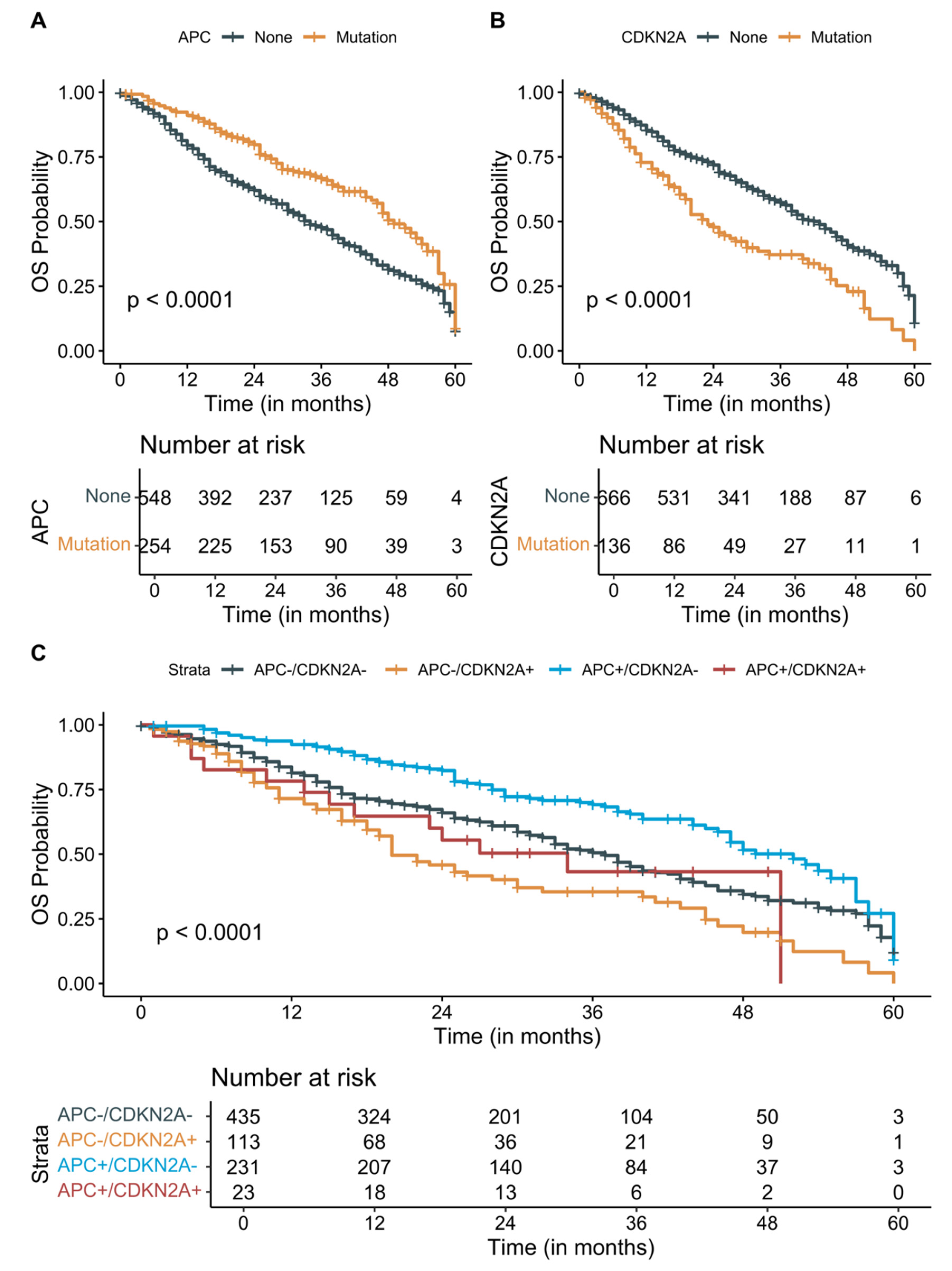
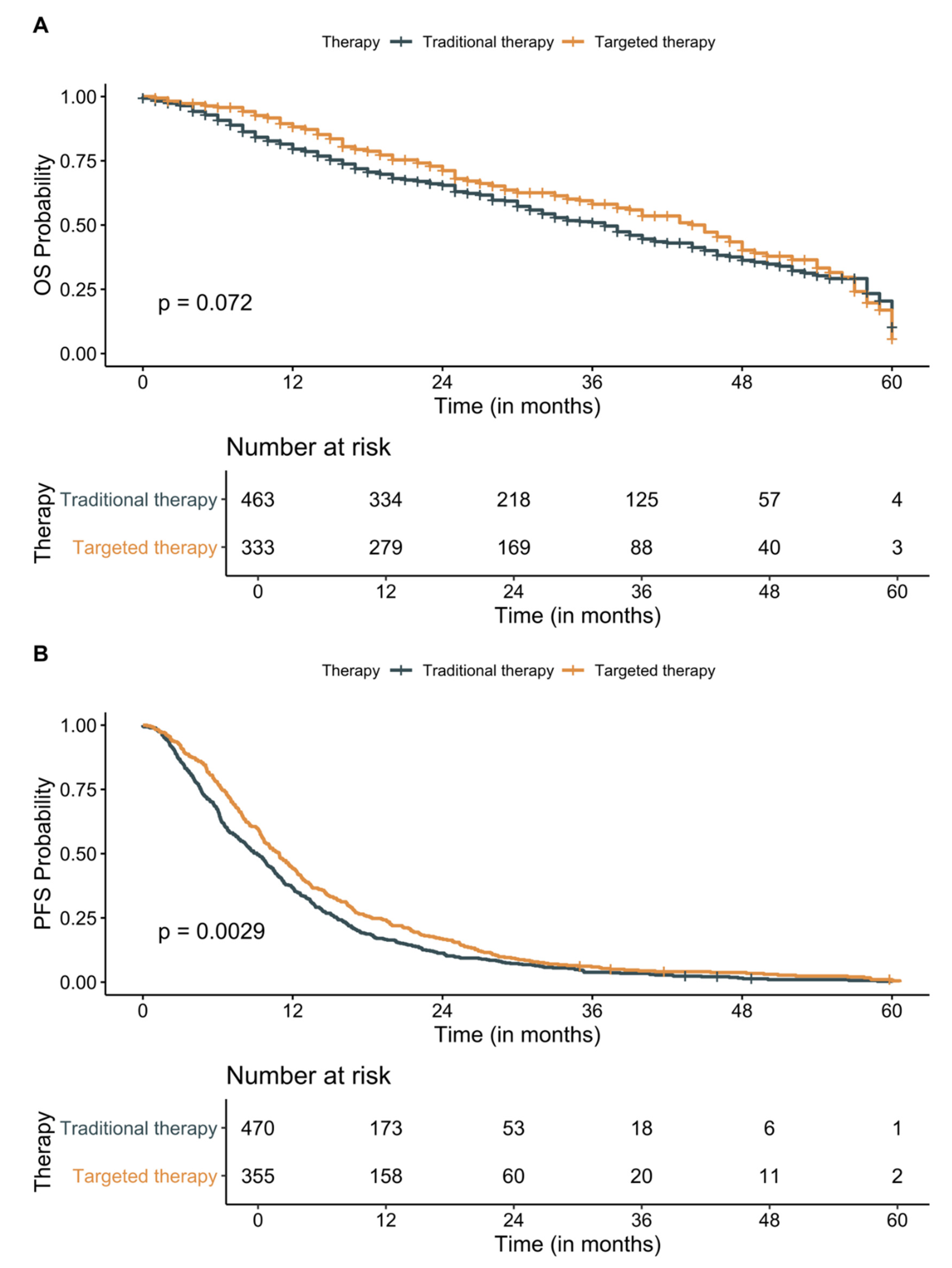
3.3. Survival Analysis
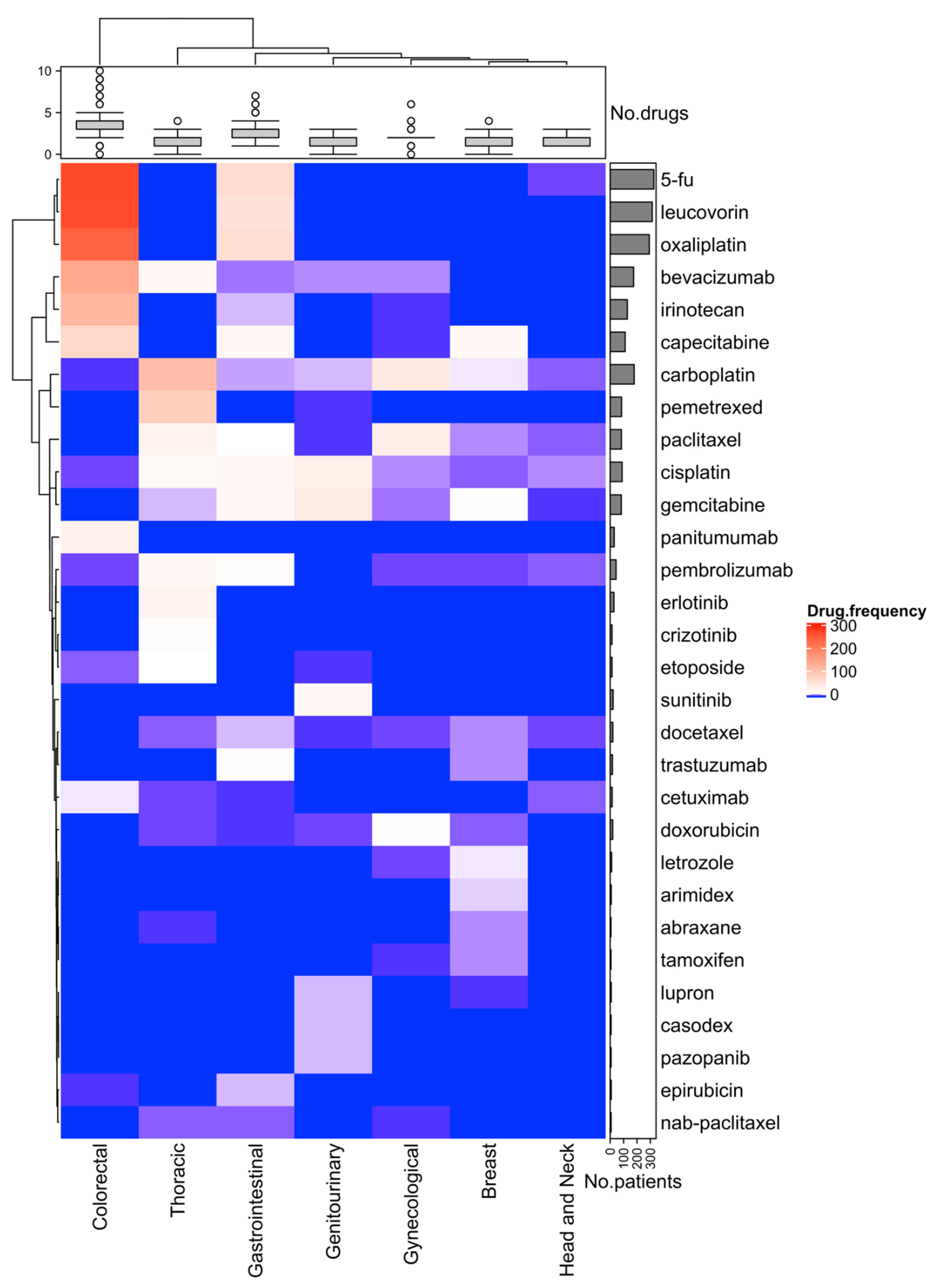
3.4. Effect of Molecular Testing and First-Line Treatments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.T.; Gray, R.J.; Chen, A.P.; Li, S.; McShane, L.M.; Patton, D.; Hamilton, S.R.; Williams, P.M.; Iafrate, A.J.; Sklar, J.; et al. Molecular Landscape and Actionable Alterations in a Genomically Guided Cancer Clinical Trial: National Cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH). J. Clin. Oncol. 2020, 38, 3883–3894. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef] [Green Version]
- Vaz, M.; Hwang, S.Y.; Kagiampakis, I.; Phallen, J.; Patil, A.; O’Hagan, H.; Murphy, L.; Zahnow, C.A.; Gabrielson, E.; Velculescu, V.E.; et al. Chronic Cigarette Smoke-Induced Epigenomic Changes Precede Sensitization of Bronchial Epithelial Cells to Single-Step Transformation by KRAS Mutations. Cancer Cell 2017, 32, 360–376.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nat. Cell Biol. 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Petljak, M.; Alexandrov, L.B. Understanding mutagenesis through delineation of mutational signatures in human cancer. Carcinogenesis 2016, 37, 531–540. [Google Scholar] [CrossRef] [Green Version]
- Hayward, N.; Wilmott, J.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.-M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nat. Cell Biol. 2017, 545, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nat. Cell Biol. 2009, 458, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.R.; Wu, Y.-M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative clinical genomics of metastatic cancer. Nat. Cell Biol. 2017, 548, 297–303. [Google Scholar] [CrossRef]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skidmore, Z.; Wagner, A.H.; Lesurf, R.; Campbell, K.M.; Kunisaki, J.; Griffith, O.; Griffith, M. GenVisR: Genomic Visualizations in R. Bioinformatics 2016, 32, 3012–3014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canisius, S.; Martens, J.W.M.; Wessels, L.F.A. A novel independence test for somatic alterations in cancer shows that biology drives mutual exclusivity but chance explains most co-occurrence. Genome Biol. 2016, 17, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Chen, K.-Z.; Hui, B.-G.; Zhang, K.; Yang, F.; Wang, J. Role of circulating tumor DNA in the management of early-stage lung cancer. Thorac. Cancer 2018, 9, 509–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassambara, A.K.; Biecek, M. P Survminer: Drawing Survival Curves Using ’ggplot2′, R package version 0.4.6; 2019. Available online: https://rpkgs.datanovia.com/survminer/index.html (accessed on 8 August 2020).
- Therneau, T. A Package for Survival Analysis in R, R package version 3.1-12; 2020. Available online: https://cran.r-project.org/web/packages/survival/index.html (accessed on 8 July 2020).
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- NCCN® Clinical Practical Guidelines in Oncology. Available online: https://www.nccn.org/professionals/physician_gls/default.aspx (accessed on 13 December 2020).
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Yang, T.-H.O.; Leiserson, M.D.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform Analysis of 12 Cancer Types Reveals Molecular Classification within and across Tissues of Origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, P.J.; Getz, G.; Korbel, J.O.; Stuart, J.M.; Jennings, J.L.; Stein, L.D.; Perry, M.D.; Nahal-Bose, H.K.; Ouellette, B.F.F.; Li, C.H.; et al. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, Y.; Varambally, S.; Creighton, C.J. Molecular Correlates of Metastasis by Systematic Pan-Cancer Analysis Across the Cancer Genome Atlas. Mol. Cancer Res. 2019, 17, 476–487. [Google Scholar] [CrossRef] [Green Version]
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.-K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 1370–1384.e5. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Shay, J.W. Multiple Roles of APC and its Therapeutic Implications in Colorectal Cancer. J. Natl. Cancer Inst. 2017, 109, djw332. [Google Scholar] [CrossRef] [Green Version]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Chong, W.; Wu, Q.; Yao, Y.; Mao, M.; Wang, X. Association of LRP1B Mutation with Tumor Mutation Burden and Outcomes in Melanoma and Non-small Cell Lung Cancer Patients Treated with Immune Check-Point Blockades. Front. Immunol. 2019, 10, 1113. [Google Scholar] [CrossRef] [Green Version]
- Fodde, R.; Kuipers, J.; Rosenberg, C.; Smits, R.; Kielman, M.; Gaspar, C.; Van Es, J.H.; Breukel, C.; Wiegant, J.; Giles, R.H.; et al. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat. Cell Biol. 2001, 3, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ouyang, C.; Cho, M.; Ji, J.; Sandhu, J.; Goel, A.; Kahn, M.; Fakih, M. Wild-type APC Is Associated with Poor Survival in Metastatic Microsatellite Stable Colorectal Cancer. Oncologist 2021, 26, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Schell, M.J.; Yang, M.; Teer, J.K.; Lo, F.Y.; Madan, A.; Coppola, D.; Monteiro, A.N.A.; Nebozhyn, M.V.; Yue, B.; Loboda, A.; et al. A multigene mutation classification of 468 colorectal cancers reveals a prognostic role for APC. Nat. Commun. 2016, 7, 11743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorissen, R.N.; Christie, M.; Mouradov, D.; Sakthianandeswaren, A.; Li, S.; Love, C.; Xu, Z.-Z.; Molloy, P.L.; Jones, I.T.; McLaughlin, S.; et al. Wild-type APC predicts poor prognosis in microsatellite-stable proximal colon cancer. Br. J. Cancer 2015, 113, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochoa, S.; Martínez-Pérez, E.; Zea, D.J.; Molina-Vila, M.A.; Marino-Buslje, C. Co-mutation and exclusion analysis in human tumors, a means for cancer biology studies and treatment design. bioRxiv 2017, 182501. [Google Scholar] [CrossRef] [Green Version]
- Bae, N.C.; Chae, M.H.; Lee, M.H.; Kim, K.M.; Lee, E.B.; Kim, C.H.; Park, T.-I.; Han, S.B.; Jheon, S.; Jung, T.H.; et al. EGFR, ERBB2, and KRAS mutations in Korean non-small cell lung cancer patients. Cancer Genet. Cytogenet. 2007, 173, 107–113. [Google Scholar] [CrossRef]
- Bradshaw, R.; Chalkley, R.; Biarc, J.; Burlingame, A.L. Receptor tyrosine kinase signaling mechanisms: Devolving TrkA responses with phosphoproteomics. Adv. Biol. Regul. 2013, 53, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, T.L.; Lertpiriyapong, K.; Cocco, L.; Martelli, A.M.; Libra, M.; Candido, S.; Montalto, G.; Cervello, M.; Steelman, L.; Abrams, S.L.; et al. Roles of EGFR and KRAS and their downstream signaling pathways in pancreatic cancer and pancreatic cancer stem cells. Adv. Biol. Regul. 2015, 59, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Ju, L.; Han, M.; Zhao, C.; Li, X. EGFR, KRAS and ROS1 variants coexist in a lung adenocarcinoma patient. Lung Cancer 2016, 95, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Benesova, L.; Minarik, M.; Jancarikova, D.; Belsanova, B.; Pesek, M. Multiplicity of EGFR and KRAS mutations in non-small cell lung cancer (NSCLC) patients treated with tyrosine kinase inhibitors. Anticancer Res. 2010, 30, 1667–1671. [Google Scholar] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szymańska, K.; Moore, L.; Rothman, N.; Chow, W.; Waldman, F.; Jaeger, E.; Waterboer, T.; Foretova, L.; Navrátilová, M.; Janout, V.; et al. TP53, EGFR, and KRAS mutations in relation to VHL inactivation and lifestyle risk factors in renal-cell carcinoma from central and eastern Europe. Cancer Lett. 2010, 293, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Ko, T.K.; Kelly, E.; Pines, J. CrkRS: A novel conserved Cdc2-related protein kinase that colocalises with SC35 speckles. J. Cell Sci. 2001, 114, 2591–2603. [Google Scholar] [CrossRef]
- Paculova, H.; Kohoutek, J. The emerging roles of CDK12 in tumorigenesis. Cell Div. 2017, 12, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertins, P.; Cptac, N.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nat. Cell Biol. 2016, 534, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capra, M.; Nuciforo, P.G.; Confalonieri, S.; Quarto, M.; Bianchi, M.; Nebuloni, M.; Boldorini, R.; Pallotti, F.; Viale, G.; Gishizky, M.L.; et al. Frequent Alterations in the Expression of Serine/Threonine Kinases in Human Cancers. Cancer Res. 2006, 66, 8147–8154. [Google Scholar] [CrossRef] [Green Version]
- Pilarova, K.; Herudek, J.; Blazek, D. CDK12: Cellular functions and therapeutic potential of versatile player in cancer. NAR Cancer 2020, 2, zcaa003. [Google Scholar] [CrossRef] [Green Version]
- Sircoulomb, F.; Bekhouche, I.; Finetti, P.; Adélaïde, J.; Ben Hamida, A.; Bonansea, J.; Raynaud, S.; Innocenti, C.; Charafe-Jauffret, E.; Tarpin, C.; et al. Genome profiling of ERBB2-amplified breast cancers. BMC Cancer 2010, 10, 539. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.R.; Udyavar, A.R.; Chang, C.-W.; Spoerke, J.M.; Aimi, J.; Savage, H.M.; Daemen, A.; O’Shaughnessy, J.A.; Bourgon, R.; Lackner, M.R.; et al. Genomic Alterations Associated with Recurrence and TNBC Subtype in High-Risk Early Breast Cancers. Mol. Cancer Res. 2018, 17, 97–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papillon-Cavanagh, S.; Doshi, P.; Dobrin, R.; Szustakowski, J.; Walsh, A.M. STK11 and KEAP1 mutations as prognostic biomarkers in an observational real-world lung adenocarcinoma cohort. ESMO Open 2020, 5, e000706. [Google Scholar] [CrossRef] [Green Version]
- Morganti, S.; Tarantino, P.; Ferraro, E.; D’Amico, P.; Duso, B.A.; Curigliano, G. Next Generation Sequencing (NGS): A Revolutionary Technology in Pharmacogenomics and Personalized Medicine in Cancer. Adv. Exp. Med. Biol. 2019, 1168, 9–30. [Google Scholar] [CrossRef]
- Postel, M.; Roosen, A.; Laurent-Puig, P.; Taly, V.; Wang-Renault, S.-F. Droplet-based digital PCR and next generation sequencing for monitoring circulating tumor DNA: A cancer diagnostic perspective. Expert Rev. Mol. Diagn. 2018, 18, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Tan, O.; Shrestha, R.; Cunich, M.; Schofield, D. Application of next-generation sequencing to improve cancer management: A review of the clinical effectiveness and cost-effectiveness. Clin. Genet. 2018, 93, 533–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groisberg, R.; Hong, D.S.; Roszik, J.; Janku, F.; Tsimberidou, A.M.; Javle, M.; Meric-Bernstam, F.; Subbiah, V. Clinical Next-Generation Sequencing for Precision Oncology in Rare Cancers. Mol. Cancer Ther. 2018, 17, 1595–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zehir, A.; Benayed, R.; Shah, R.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Toomey, S.; Carr, A.; Mezynski, M.J.; Elamin, Y.; Rafee, S.; Cremona, M.; Morgan, C.; Madden, S.; Abdul-Jalil, K.I.; Gately, K.; et al. Identification and clinical impact of potentially actionable somatic oncogenic mutations in solid tumor samples. J. Transl. Med. 2020, 18, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Kim, N.K.D.; Lee, S.H.; Cho, H.W.; Ma, Y.; Ju, H.Y.; Yoo, K.H.; Sung, K.W.; Koo, H.H.; Park, W.-Y. Discovery of actionable genetic alterations with targeted panel sequencing in children with relapsed or refractory solid tumors. PLoS ONE 2019, 14, e0224227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drilon, A.; Wang, L.; Arcila, M.E.; Balasubramanian, S.; Greenbowe, J.R.; Ross, J.S.; Stephens, P.J.; Lipson, D.; Miller, V.A.; Kris, M.G.; et al. Broad, Hybrid Capture–Based Next-Generation Sequencing Identifies Actionable Genomic Alterations in Lung Adenocarcinomas Otherwise Negative for Such Alterations by Other Genomic Testing Approaches. Clin. Cancer Res. 2015, 21, 3631–3639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, G.A.; Miller, V.A.; Curran, J.; Ross, J.S.; Lipson, D.; Yelensky, R.; Stephens, P.; Lancelotta, M.P.; Cronin, M.T. Next-generation sequencing (NGS) to identify actionable genomic changes in common and rare solid tumors: The FMI experience with the initial 50 consecutive patients. J. Clin. Oncol. 2012, 30, 10590. [Google Scholar] [CrossRef]
- Miller, V.A.; Ross, J.S.; Wang, K.; Ali, S.M.; Otto, G.; Curran, J.; Palma, N.A.; Yelensky, R.; Downing, S.; Stephens, P.; et al. Use of next-generation sequencing (NGS) to identify actionable genomic alterations (GA) in diverse solid tumor types: The Foundation Medicine (FMI) experience with 2200+ clinical samples. J. Clin. Oncol. 2013, 31, 11020. [Google Scholar] [CrossRef]
- Hainsworth, J.D.; Meric-Bernstam, F.; Swanton, C.; Hurwitz, H.; Spigel, D.R.; Sweeney, C.; Burris, H.A.; Bose, R.; Yoo, B.; Stein, A.; et al. Targeted Therapy for Advanced Solid Tumors on the Basis of Molecular Profiles: Results From MyPathway, an Open-Label, Phase IIa Multiple Basket Study. J. Clin. Oncol. 2018, 36, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.-M.; Hong, D.S.; Ye, Y.; Cartwright, C.; Wheler, J.J.; Falchook, G.S.; Naing, A.; Fu, S.; Piha-Paul, S.; Janku, F.; et al. Initiative for Molecular Profiling and Advanced Cancer Therapy (IMPACT): An MD Anderson Precision Medicine Study. JCO Precis. Oncol. 2017, 2017, 1–18. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Hong, D.S.; Kuo, J.; Sacher, A.G.; Barlesi, F.; Besse, B.; Kuboki, Y.; Dy, G.K.; Dembla, V.; Krauss, J.C.; Burns, T.F.; et al. CodeBreak 100: Phase I study of AMG 510, a novel KRASG12C inhibitor, in patients (pts) with advanced solid tumors other than non-small cell lung cancer (NSCLC) and colorectal cancer (CRC). J. Clin. Oncol. 2020, 38, 3511. [Google Scholar] [CrossRef]
- Wolf, J.; Seto, T.; Han, J.-Y.; Reguart, N.; Garon, E.B.; Groen, H.J.; Tan, D.S.; Hida, T.; de Jonge, M.; Orlov, S.V.; et al. Capmatinib inMETExon 14–Mutated orMET-Amplified Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Oxnard, G.R.; Tan, D.S.; Loong, H.H.; Johnson, M.; Gainor, J.; McCoach, C.E.; Gautschi, O.; Besse, B.; Cho, B.C.; et al. Efficacy of Selpercatinib in RET Fusion–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 813–824. [Google Scholar] [CrossRef]
- Rahman, M.; MacNeil, S.M.; Jenkins, D.F.; Shrestha, G.; Wyatt, S.R.; McQuerry, J.A.; Piccolo, S.R.; Heiser, L.M.; Gray, J.W.; Johnson, W.E.; et al. Activity of distinct growth factor receptor network components in breast tumors uncovers two biologically relevant subtypes. Genome Med. 2017, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Bennett, R.L.; Licht, J.D. Targeting Epigenetics in Cancer. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 187–207. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Murthy, R.K.; Loi, S.; Okines, A.; Paplomata, E.; Hamilton, E.; Hurvitz, S.A.; Lin, N.U.; Borges, V.; Abramson, V.; Anders, C.; et al. Tucatinib, Trastuzumab, and Capecitabine for HER2-Positive Metastatic Breast Cancer. N. Engl. J. Med. 2020, 382, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Bauer, T.M.; De Marinis, F.; Felip, E.; Goto, Y.; Liu, G.; Mazieres, J.; Kim, D.-W.; Mok, T.; Polli, A.; et al. First-Line Lorlatinib or Crizotinib in Advanced ALK-Positive Lung Cancer. N. Engl. J. Med. 2020, 383, 2018–2029. [Google Scholar] [CrossRef]
- De Bono, J.S.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E–Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheco, J.M.; Gao, D.; Smith, D.; Purcell, T.; Hancock, M.; Bunn, P.; Robin, T.; Liu, A.; Karam, S.; Gaspar, L.; et al. Natural History and Factors Associated with Overall Survival in Stage IV ALK-Rearranged Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2019, 14, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Kochanowski, K.; Morinishi, L.; Altschuler, S.J.; Wu, L.F. Drug persistence—From antibiotics to cancer therapies. Curr. Opin. Syst. Biol. 2018, 10, 1–8. [Google Scholar] [CrossRef]
- Levin-Reisman, I.; Brauner, A.; Ronin, I.; Balaban, N.Q. Epistasis between antibiotic tolerance, persistence, and resistance mutations. Proc. Natl. Acad. Sci. USA 2019, 116, 14734–14739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizrahi, S.P.; Gefen, O.; Simon, I.; Balaban, N.Q. Persistence to anti-cancer treatments in the stationary to proliferating transition. Cell Cycle 2016, 15, 3442–3453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaban, N.Q.; Helaine, S.; Lewis, K.; Ackermann, M.; Aldridge, B.; Andersson, D.I.; Brynildsen, M.P.; Bumann, D.; Camilli, A.; Collins, J.J.; et al. Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Genet. 2019, 17, 441–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Pan-Cancer (n = 957) | Breast (n = 87) | Colorectal (n = 322) | Gastrointestinal (n = 123) | Genitourinary (n = 118) | Gynecological (n = 59) | Head and Neck (n = 19) | Thoracic (n = 229) |
|---|---|---|---|---|---|---|---|---|
| Age at diagnosis, median (IQR) | 60 (50–69) | 50 (42–56) | 56 (48–66) | 63 (49–70) | 67 (60–73) | 61 (51.5–71) | 61 (50.5–65.5) | 64 (54–72) |
| Race, n (%) | ||||||||
| White | 667 (69.7) | 50 (57.5) | 230 (71.4) | 74 (60.2) | 98 (83.1) | 47 (79.7) | 17 (89.5) | 151 (65.9) |
| Asian | 194 (20.3) | 18 (20.7) | 60 (18.6) | 35 (28.5) | 13 (11) | 8 (13.6) | 1 (5.3) | 59 (25.9) |
| African American | 47 (4.9) | 12 (13.8) | 15 (4.7) | 4 (3.3) | 2 (1.7) | 2 (3.4) | 1 (5.3) | 11 (4.8) |
| Other | 18 (1.9) | 4 (4.6) | 7 (2.1) | 7 (5.7) | 3 (2.5) | 2 (3.7) | 0 (0) | 4 (1.7) |
| Unknown | 22 (2.3) | 3 (3.5) | 10 (3.1) | 3 (2.4) | 2 (1.6) | 0 (0) | 0(0) | 4 (1.7) |
| Ethnicity, n (%) | ||||||||
| Hispanic or Latino | 193 (20.2) | 24 (27.6) | 74 (23) | 37 (30.1) | 10 (8.5) | 16 (27.1) | 3 (15.8) | 29 (12.7) |
| Not Hispanic or Latino | 752 (78.6) | 61(70.1) | 244 (75.8) | 83 (67.5) | 108 (91.5) | 43 (72.9) | 16 (84.2) | 197 (86) |
| Unknown or not disclosed | 12 (1.2) | 2(2.3) | 4 (1.2) | 3 (2.4) | 0 (0) | 0 (0) | 0 (0) | 3 (1.3) |
| Sex, n (%) | ||||||||
| Female | 497 (51.9) | 87 (100) | 138 (42.9) | 54 (43.9) | 27 (22.9) | 59 (100) | 8 (42.1) | 124 (54.1) |
| Male | 460 (48.1) | 0 (0) | 184 (57.1) | 69 (56.1) | 91 (77.1) | 0 (0) | 11 (57.9) | 105 (45.9) |
| Tumor Burden, n (%) | ||||||||
| Low | 204 (21.2) | 19 (21.8) | 70 (21.7) | 20 (16.3 | 16 (13.1) | 21 (35.6) | 10 (52.6) | 48 (21.1) |
| Intermediate | 115 (12) | 6 (6.9) | 37 (11.5) | 17 (13.8) | 8 (6.6) | 10 (16.9) | 3 (15.8) | 34 (14.9) |
| High | 17 (1.8) | 0 (0) | 2 (0.6) | 1 (0.8) | 1 (0.8) | 1 (1.7) | 2 (10.5) | 10 (4.4) |
| Unknown | 624 (65) | 62 (71.2) | 221 (66.1) | 85 (69.1) | 97 (79.5) | 27 (45.8) | 4 (21.1) | 136 (59.6) |
| Stage at initial diagnosis, n (%) | ||||||||
| I | 41 (4.3) | 16 (18.4) | 9 (2.8) | 0 (0) | 0 (0) | 5 (8.5) | 1 (5.3) | 10 (4.4) |
| II | 81 (8.5) | 32 (36.8) | 29 (9) | 2 (1.6) | 0 (0) | 4 (6.8) | 3 (15.8) | 11 (4.8) |
| III | 134 (14) | 23 (26.4) | 62 (19.3) | 3 (2.4) | 4 (3.9) | 23 (39) | 0 (0) | 19 (8.3) |
| IV | 701 (73.3) | 16 (18.4) | 222 (68.9) | 118 (95.9) | 114 (96.6) | 27 (45.8) | 15 (78.9) | 189 (82.5) |
| MSI status, n (%) | ||||||||
| High | 8 (0.8) | 0 (0) | 2 (0.6) | 2 (1.6) | 0 (0) | 1 (1.7) | 1 (5.3) | 2 (0.9) |
| Stable | 351 (36.7) | 26 (29.9) | 116 (36) | 39 (31.7) | 25 (21.2) | 32 (54.8) | 15 (78.9) | 98 (42.8) |
| Unknown | 598 (62.5) | 60 (70.1) | 204 (63.4) | 82 (66.7) | 93 (78.8) | 26 (44.1) | 3 (15.8) | 129 (56.3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roosan, M.R.; Mambetsariev, I.; Pharaon, R.; Fricke, J.; Baroz, A.R.; Chao, J.; Chen, C.; Nasser, M.W.; Chirravuri-Venkata, R.; Jain, M.; et al. Evaluation of Somatic Mutations in Solid Metastatic Pan-Cancer Patients. Cancers 2021, 13, 2776. https://doi.org/10.3390/cancers13112776
Roosan MR, Mambetsariev I, Pharaon R, Fricke J, Baroz AR, Chao J, Chen C, Nasser MW, Chirravuri-Venkata R, Jain M, et al. Evaluation of Somatic Mutations in Solid Metastatic Pan-Cancer Patients. Cancers. 2021; 13(11):2776. https://doi.org/10.3390/cancers13112776
Chicago/Turabian StyleRoosan, Moom R., Isa Mambetsariev, Rebecca Pharaon, Jeremy Fricke, Angel R. Baroz, Joseph Chao, Chen Chen, Mohd W. Nasser, Ramakanth Chirravuri-Venkata, Maneesh Jain, and et al. 2021. "Evaluation of Somatic Mutations in Solid Metastatic Pan-Cancer Patients" Cancers 13, no. 11: 2776. https://doi.org/10.3390/cancers13112776