Multiple Intraspecific Variations of Mitochondrial Genomes in the Green-Tide Forming Alga, Ulva compressa Linnaeus (Ulvophyceae, Chlorophyta)
 Feng Liu
Feng Liu James T. Melton III
James T. Melton III Juan M. Lopez-Bautista
Juan M. Lopez-Bautista Nansheng Chen
Nansheng Chen- 1CAS Key Laboratory of Marine Ecology and Environmental Sciences, Institute of Oceanology, Chinese Academy of Sciences (IOCAS), Qingdao, China
- 2Marine Ecology and Environmental Science Laboratory, Pilot National Laboratory for Marine Science and Technology (Qingdao), Qingdao, China
- 3Center for Ocean Mega-Science, Chinese Academy of Sciences, Qingdao, China
- 4Spelman College, Atlanta, GA, United States
- 5Department of Biological Sciences, The University of Alabama, Tuscaloosa, AL, United States
To gain further insights into intraspecific evolution of Ulva mitochondrial genomes, the mitogenomes of three morphotypes of the green-tide forming alga, Ulva compressa Linnaeus from China and United States, were sequenced and compared with the available data from Ulvophyceae. The U. compressa mitogenomes displayed substantial genome size variation at intraspecific level ranging from 61,700 to > 66,587 bp, due to different acquisitions of foreign DNA fragments, gain or loss of both group I and II introns, and non-coding intergenic spacer regions. The U. compressa mitogenomes harbored variable gene content ranging from 69 genes (including orfs) in Uco1 to 76 in Uco5, and contained different intron content from 4 introns in Uco3 and Uco4 to 7 in Uco1. A total of 63 genes and only two group IB introns (intron cox1-1107 and cox1-1125) were shared by these five mitogenomes. The U. compressa mitogenomes accumulated many more inverted repeat (IR) elements, ranging from 45 in Uco1 to 88 in Uco2, than that of the other Ulva species (3–34). A locally collinear block of eight genes (rps11-rps19-rps4-rpl16-trnR-trnQ-trnE-trnS) with the size of 3,631 bp has been inverted only in Uco1 indicating that the rearrangement event happened after its divergence from Uco2–5 and might be related to a specific IR element. The majority of the common genes (~76%) displayed high identity (>98%) among these five mitogenomes, while some low values were observed in six genes mainly due to duplication and insertion/deletion mutations of small DNA fragments. Our study presented the first case of multiple intraspecific variations in ulvophycean mitogenomes, and indicated that the mitogenome will be a valuable tool for understanding the native or non-indigenous nature of the cosmopolitan Ulva species.
Introduction
The class Ulvophyceae is one of the five classes of green algae in the core Chlorophyta (Burger and Nedelcu, 2012; Leliaert et al., 2012; Fuèíková et al., 2014), and contains ten orders and more than 1,900 species thus far (Guiry and Guiry, 2020). Species in Ulvophyceae have developed four main cytomorphological types including non-motile uninucleate unicells, multicellular filaments or blades consisting of uninucleate cells, multicellular thalli composed of multinucleate cells, and coenocytic thalli (Mine et al., 2008; Cocquyt et al., 2010; Leliaert et al., 2012). Phylogenetics is required to develop a comprehensive inventory of the Ulvophyceae to understand the evolutionary history and biodiversity over time. Mitochondrial genomes (mitogenomes) have been useful in phylogenetics, phylogeography and population genetic studies at various taxonomic levels due to the high mutation rates, abundance in cells, and genome-level content (Gray et al., 2001; Hanyuda et al., 2016).
Despite the rich biodiversity and various morphological types in Ulvophyceae, the mitochondrial genomes have been sequenced for only 16 ulvophycean taxa including nine in the order Ulvales (all Ulva spp.), three in Ulotrichales, three in Bryopsidales, and one in Oltmannsiellopsidales (Table 1). The reported ulvophycean mitogenomes exhibit large variation in genome size, architechtures, gene density, and intron content (Liu et al., 2017). Species in Bryopsidales harbor the inflated mitogenomes ranged from 197,427 bp in Caulerpa ashmeadii to 241,739 bp in Ostreobium quekettii, due to expansion of non-coding DNA and proliferation of introns (Sauvage et al., 2019; Repetti et al., 2020). Species in Ulotrichales tend to contain large mitogenomes, such as the 105,236-bp Gloeotilopsis planctonica and 95,880-bp Pseudendoclonium akinetum mitogenomes, which also display characteristics of the ‘expanded-derived’ patterns of evolution (Pombert et al., 2004; Turmel et al., 2016). The reported Ulva mitogenomes ranged from 61,614 bp in Ulva lactuca to 73,493 bp in Ulva sp. UNA00071828, which were smaller than that of Bryopsidales and Ulotrichales (Melton et al., 2015; Liu et al., 2017; Suzuki et al., 2018). The 56,761-bp mitogenome of Oltmannsiellopsis viridis (Oltmannsiellopsidales) is the smallest one reported in Ulvophyceae so far (Pombert et al., 2006).
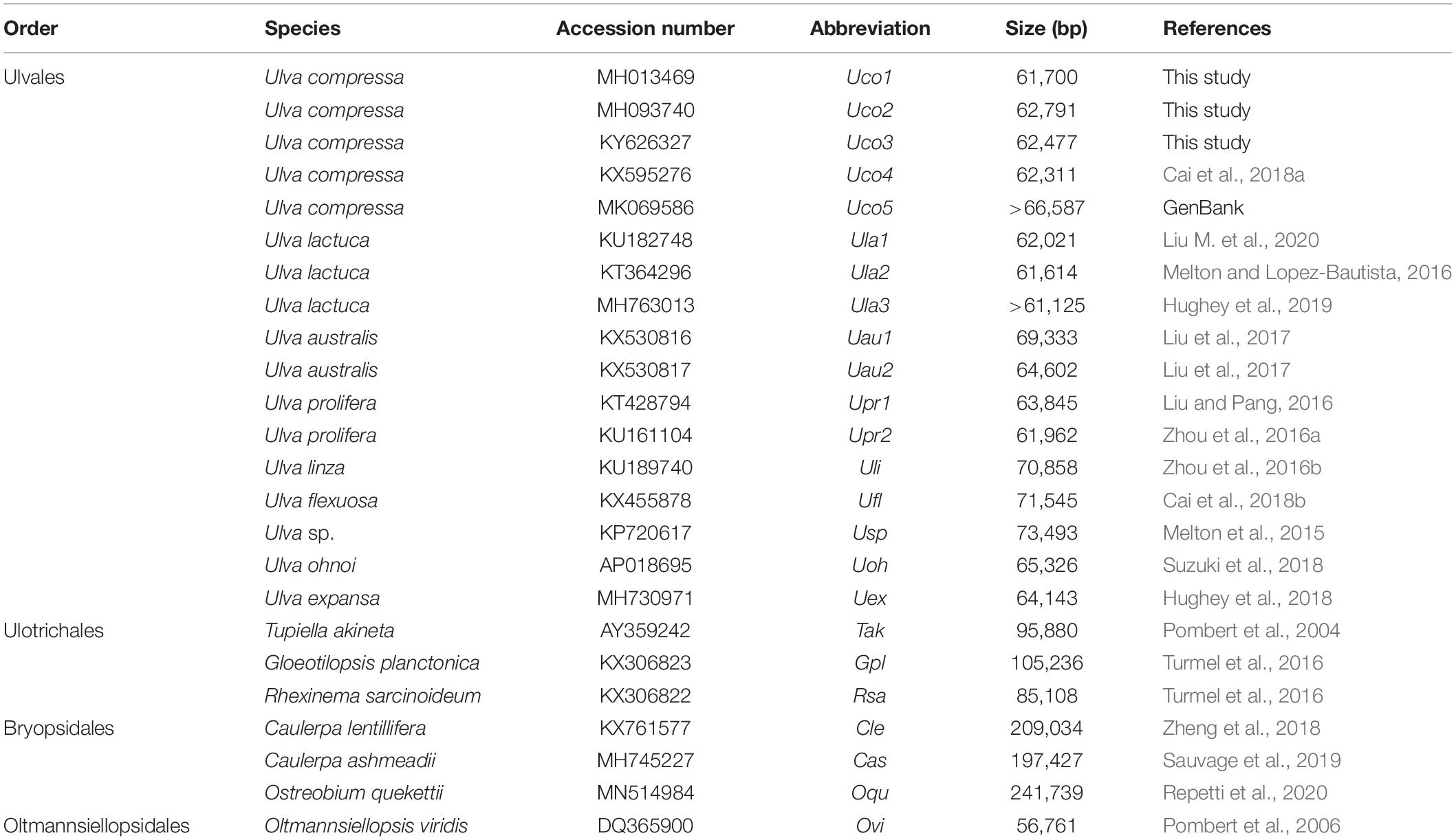
Table 1. The known 24 mitochondrial genomes from 16 ulvophycean taxa in the class Ulvophyceae.
The reported mitogenomes of nine Ulva species were conserved in genome arrangement, gene order and content of core genes, while they displayed intraspecific variation in genome size, due to intron gain or loss, insertion of foreign DNA fragments, and changes of repeat regions (Liu et al., 2017; Liu M. et al., 2020). Two mitogenomes of Ulva pertusa, which is currently regarded as a taxonomic synonym of Ulva australis, exhibited intraspecific variation of intron content, due to the insertion of two group II introns in the larger one, and the changes of tandem repeats (Liu et al., 2017). Two mitogenomes of the notorious bloom-forming alga, Ulva prolifera (Wang et al., 2019), exhibited a difference in genome size, which was mainly caused by an insertion of 1878 bp foreign DNA sequence harboring trnL(uag) and two orfs (orf253 and orf173) into the rnl-trnY(gua) intergenic spacer region (Liu and Pang, 2016). A similar insertion sequence was present in the Ulva linza mitogenome, and contained orf457 representing a fusion of orf253 and orf173 (Zhou et al., 2016a, b). Three mitogenomes of U. lactuca contain the same number of introns, although the algal thalli were sampled at three greatly distant places (Melton and Lopez-Bautista, 2016; Hughey et al., 2019), while differences in genome size among these three mitogenomes were mainly caused by duplication mutations and insertion/deletion mutations of short DNA sequences (Liu M. et al., 2020).
Ulva compressa Linnaeus has a worldwide geographic distribution in marine and estuarine environments (Guiry and Guiry, 2020). This alga is known for its rapid, proliferous growth in eutrophic conditions, even forming green tides (Blomster et al., 2002; Liu et al., 2013). U. compressa usually displays great morphological plasticity due to temperature, salinity, irradiance, wave exposure, and nutrient content (Steinhagen et al., 2019). Previously, two mitochondrial genomes of U. compressa, one complete mitogenome (GenBank accession number KX595276) from Southern Yellow Sea, China (Cai et al., 2018a), and another nearly complete mitogenome (MK069586) from China, had been sequenced. To gain further insights into the evolution of Ulva mitogenomes, in this study, three morphotypes of U. compressa samples were collected from China and the United States, and their complete mitogenomes were sequenced to compare with the available mitogenomes in Ulvophyceae.
Materials and Methods
Sample Collection and DNA Extraction
The algal thalli of three morphotypes of Ulva compressa Linnaeus were sampled from Swansboro, NC, United States (Figure 1A); Huiquan Bay, Qingdao, Shandong, China (Figure 1B); and Subei Shoal, Jiangsu, China (Figure 1C), respectively (Supplementary Table S1). The sample from the United States (Uco1) was preserved in silica gel and as a herbarium voucher, which was submitted to University of Alabama Herbarium (UNA00072686). The dried thallus was used to extract DNA with a Qiagen Plant DNA Extraction Kit (QIAGEN, Valencia, CA, United States). Two algal samples from China (Uco2 and Uco3) were kept in coolers (5–8°C) and transported to the laboratory in IOCAS within 48 h after collection. Fresh tissue from one individual algal thallus was used for DNA extraction using a Plant Genomic DNA Kit (Tiangen Biotech, Beijing, China) according to the manufacturer’s instructions. These three U. compressa samples showed different morphological features, which have been noted in this species (Supplementary Table S1). Two common DNA markers, the internal transcribed spacer DNA (ITS) region including the 5.8S rDNA gene and plastid-encoded large subunit of the ribulose 1,5-bisphosphate carboxylase gene (rbcL) (Hayden and Waaland, 2002), were used for species identification according to the protocol as previously described (Liu et al., 2010). The identification results confirmed that all of these three samples were U. compressa (Supplementary Figures S1, S2).
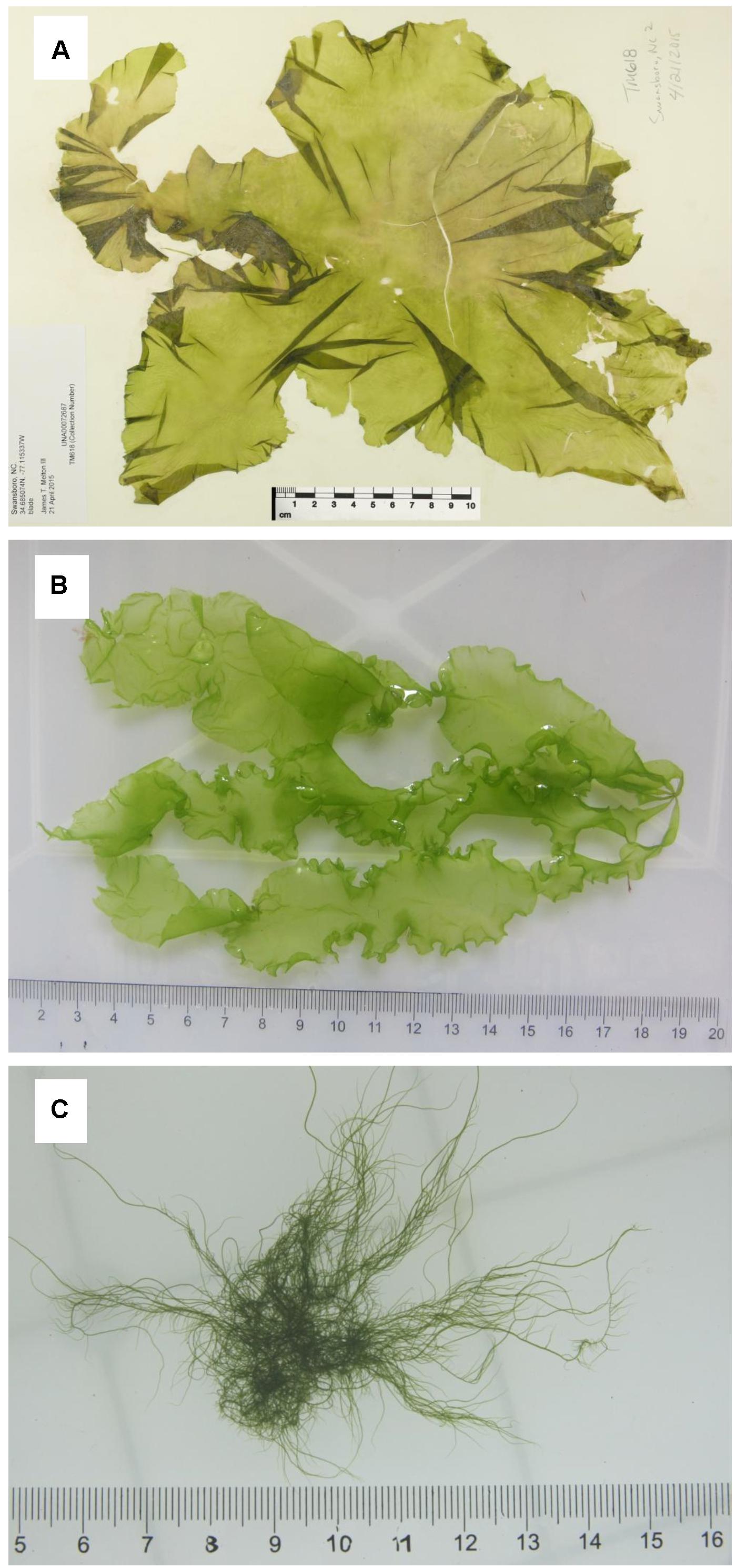
Figure 1. Three morphotypes of Ulva compressa collected from China and USA: (A) Uco1 with foliated, distromatic blade, (B) Uco2 with ribbon, distromatic blade and monostromatic, tubular margins, and (C) Uco3 with filamentous, tubular blade, and dense branches.
Sequencing and Assembly
The concentration and quality of isolated DNA were measured with a NanoDrop ND-1000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States). For the sample from the USA, paired-end reads (150 bp) were sequenced at Cold Spring Harbor Laboratory on an Illumina MiSeq platform. For two samples from China, the purified DNA was fragmented into 350 bp and used to construct short-insert libraries. The short fragments were sequenced using an Illumina Hiseq 4000 sequencing platform. The mitogenomes of U. compressa were constructed using a combination of de novo and reference-guided assemblies. Genome assembly for the sample from the USA was done with both A5 (Tritt et al., 2012) and Geneious R7 (Kearse et al., 2012). For samples from China, the filtered reads were assembled into contigs using SOAPdenovo2.04 (Luo et al., 2012). Incomplete genomes were closed by iteratively mapping the trimmed reads on to the contigs with Geneious 7.1 software1 (Supplementary Table S2).
Genome Annotation
Protein-coding genes (PCGs) were annotated by Open Reading Frame (ORF) Finder2, DOGMA(Wyman et al., 2004) and ORF finder in Geneious. The ORFs with more than 100 codons were identified in the U. compressa mitogenomes. Ribosomal RNA genes (rRNAs) were identified by a BLAST of the non-redundant databases at the National Center for Biotechnology Information (Altschul et al., 1997) and by comparing newly sequenced U. compressa mitogenomes with rRNA genes from the other ulvophyceaen mitogenomes. Transfer RNA genes (tRNAs) were searched for by reconstructing their cloverleaf structures using the tRNA scan-SE 1.21 software with default parameters (Schattner et al., 2005).
Intron Analysis
Introns were identified by aligning the homologous genes from 17 Ulva mitogenomes. We detected that some introns were not correctly annotated in the reported Ulva mitogenomes from GenBank, thus re-annotation of these introns were performed in this study. Intron insertion-sites were determined manually based on the alignments of nucleotide (nt) sequences for the homologous mitochondrial genes and/or amino acid (aa) sequences for PCGs in the mitogenome of U. compressa (KY626327) (Uco3). Intron name was defined as host gene plus insertion site. The class and core structure of all these introns were determined using the software RNAweasel (Lang et al., 2007) and Mfold (Zuker, 2003). The core domains of intronic orfs were determined by significant Pfam-A matches (Punta et al., 2012). In group IIA and IIB introns, the conserved reverse transcriptase (RT) domains with relatively complete structure were searched from the aa sequences of their intronic orfs, and 7 group IIB introns were omitted because their RT domains were degenerated or lost. Finally, the aa sequences of RT domains from 27 group IIA and IIB introns were subjected to concatenated alignments using ClustalX 1.83 with the default settings (Thompson et al., 1997). Maximum Likelihood (ML) phylogenetic tree was constructed based on the Jones et al. w/freq. model (Jones et al., 1992) with 1000 bootstrap replicates using the software MEGA 7.0 (Kumar et al., 2016). Initial trees for the heuristic search were obtained by applying the Neighbor-Joining method to a matrix of pairwise distances estimated using a JTT model. All positions containing gaps and missing data were eliminated. There were a total of 123 positions in the final dataset.
Comparative Genomic and Phylogenomic Analyses
Base composition of the 17 Ulva mitogenomes was determined using MEGA 7.0 (Kumar et al., 2016). Tandem repeats (TRs) were found with Tandem Repeats Finder using the default settings (Benson, 1999). Inverted repeats (IRs) were identified with Inverted Repeats Finder using the default settings and the additional constraint that repeats had to be > 75% similar3. A total of 61 genes including 29 core PCGs, 2 rRNAs, 27 tRNAs and 3 conserved free-standing orfs (cfORFs) were shared among the 17 Ulva mitogenomes. Differences and identity values of these gene sequences were calculated by use of the BioEdit v7.1.9 software (Hall, 1999). The nt sequences of 17 mitogenomes were subjected to concatenated alignments using ClustalX 1.83 with the default settings (Thompson et al., 1997). The evolutionary history was inferred by using the Maximum Likelihood (ML) method based on the Tamura-Nei model (Tamura and Nei, 1993). Initial trees for the heuristic search were obtained by applying the Neighbor-Joining method to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach. There were a total of 142,736 positions in the final dataset. Phylogenetic analyses were conducted with 1,000 bootstrap replicates using MEGA 7.0 (Kumar et al., 2016).
Results and Discussion
Comparison of Genome Features in U. compressa Mitogenomes
The three newly sequenced mitogenomes of U. compressa ranged from 61,700 bp in Uco1 to 62,791 bp in Uco3, which were similar to that of Uco4 in size, but were shorter than that of Uco5 (>66,587 bp) (Table 1). The variation of U. compressa mitogenome size was mainly caused by differences in the content of genome-specific free-standing orfs (sfORFs), both group I and II introns, and the non-coding intergenic spacer regions (Figure 2). The content of non-coding spacer in U. compressa mitogenomes was 22.29–26.93% (Figure 2), which was within the known range for Ulva mitogenomes (20.51–32.94%), but was lower than the mitogenomes of Bryopsidales (34.8–46.0%) (Zheng et al., 2018; Repetti et al., 2020), Ulotrichales (43.3–49.0%) (Pombert et al., 2004; Turmel et al., 2016), and Oltmannsiellopsidales (34.1%) (Pombert et al., 2006), displaying the most compact architectures among the Ulvophyceae so far.
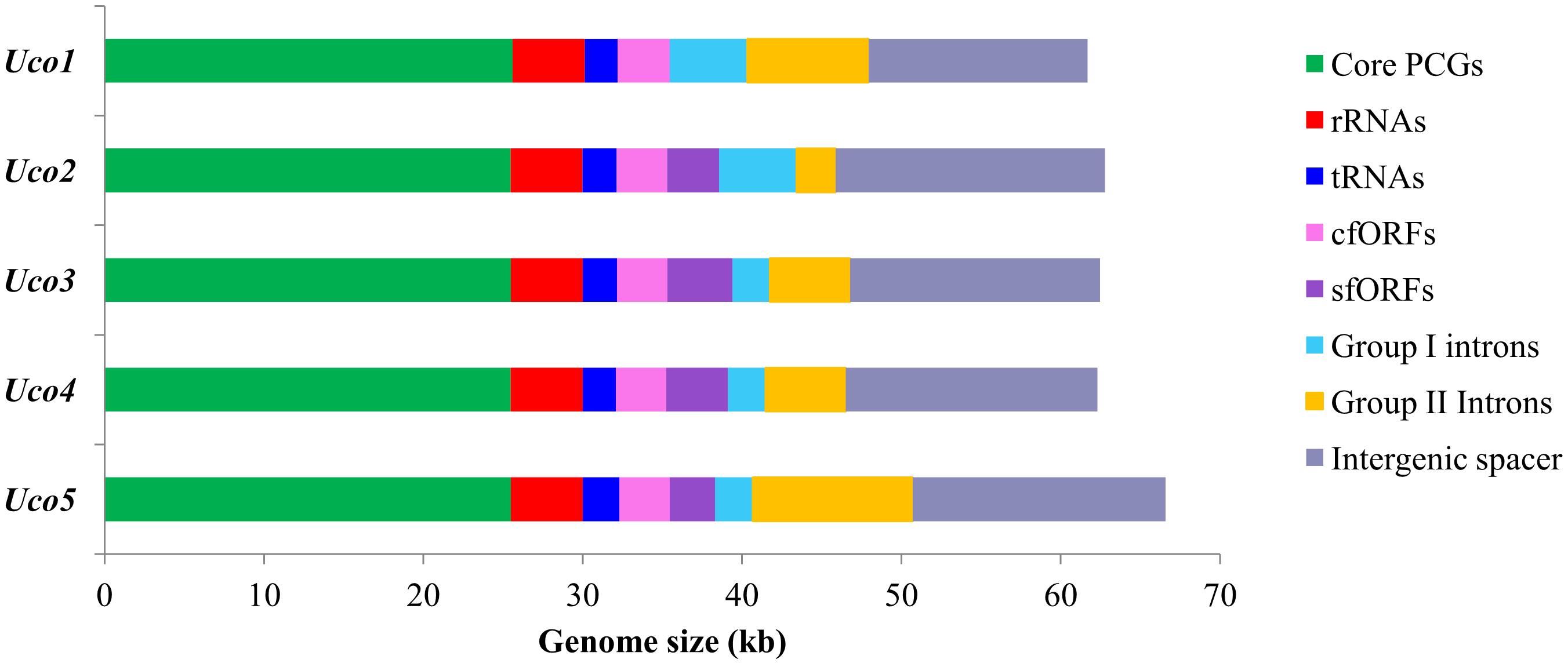
Figure 2. Contributions of 29 core PCGs, rRNA genes (rRNAs), tRNA genes (tRNAs), conserved free-standing orfs (cfORFs), genome-specific free-standing orfs (sfORFs), group I, and II introns, and non-coding intergenic spacer regions in five U. compressa mitogenomes. Horizontal bars represent the size of mitogenomes.
The overall A + T content in the United States sample (Uco1) was 61.96%, which was the lowest value recorded in Ulva mitogenomes sequenced so far (Liu et al., 2017). For each U. compressa mitogenome, the non-coding intergenic spacer regions was mostly richer in A + T content, while the A + T content in introns was much lower than that of the mitogenome. The non-coding intergenic regions tends to become richer in A + T content, as was observed in the vast majority of the mitogenomes (e.g., Joardar et al., 2012; Liu and Pang, 2015). The A + T contents in core protein-coding genes (PCGs), rRNA genes (rRNAs), tRNA genes (tRNAs) and conserved free-standing orfs (cfORFs) maintained relatively stable values among these five U. compressa mitogenomes, while the intron A + T content showed variations ranging from 55.63% in Uco5 to 61.45% in Uco2 (Table 2).
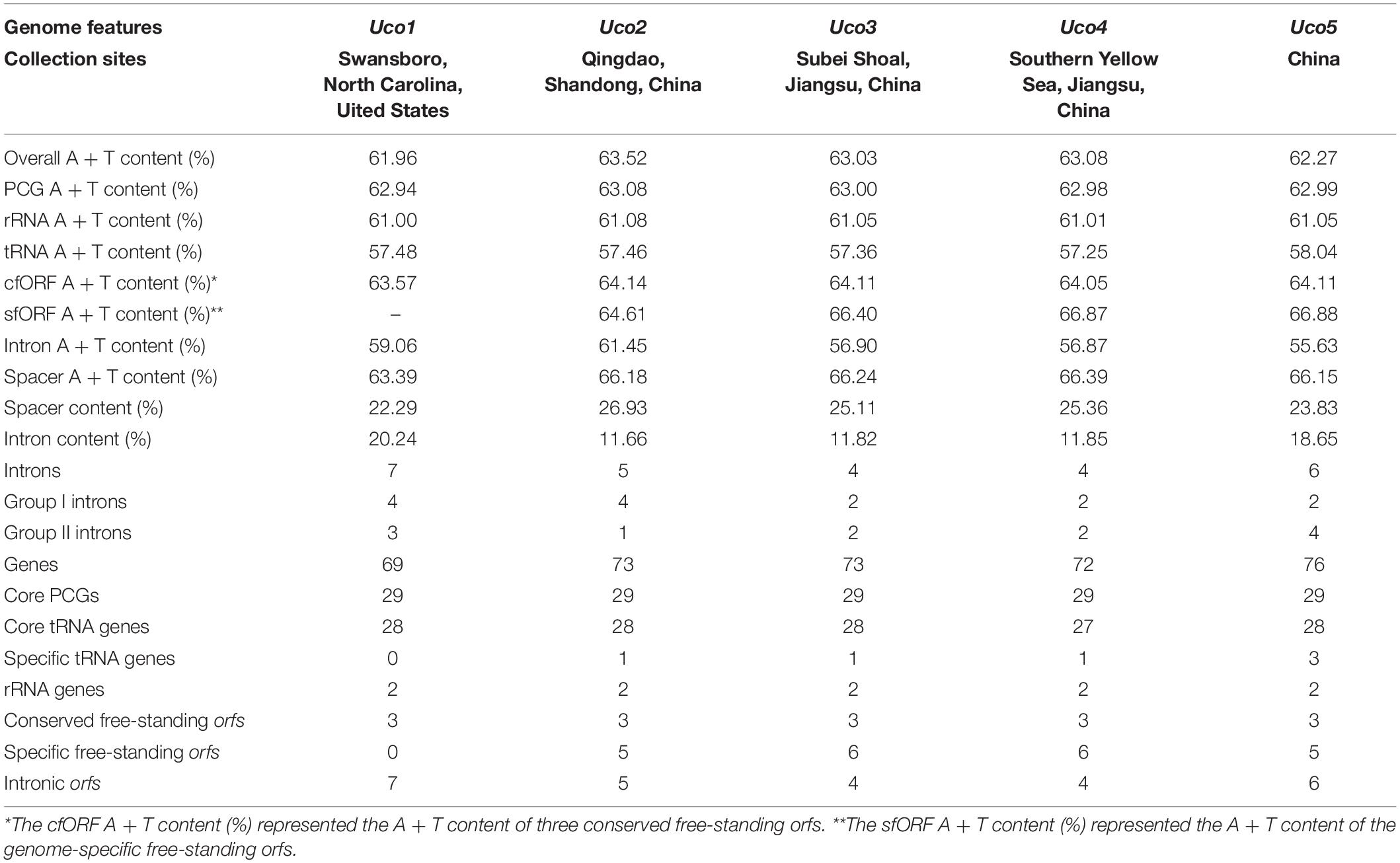
Table 2. Comparison of genome features in the five mitochondrial genomes of Ulva compressa (Uco1 – 5).
Variation of Gene Content in U. compressa Mitogenomes
The five mitogenomes of U. compressa harbored variable gene content ranging from 69 genes (including orfs) in Uco1 to 76 in Uco5 (Table 2). The differences were mainly due to the acquisition of foreign DNA fragments, which carried different numbers of genome-specific tRNAs and orfs, and gain or loss of group I and II introns which usually harbored one intronic orf. The five mitogenomes of U. compressa shared a total of 63 genes including 29 core PCGs, two rRNAs, 27 tRNAs, three cfORFs (Figure 3) and two intronic orfs situated in two group IB introns (i.e. intron cox1-1107 and cox1-1125) (Table 3). The 29 PCGs including five ATPase subunits (atp1, 4, 6, 8 and 9), apocytochrome b (cob), three cytochrome oxidase subunits (cox1-3), eight NADH dehydrogenase complex subunits (nad1-7, and 4L), and 12 ribosomal proteins (rpl5, 14, and 16; rps2-4, 10-14, and 19) were conserved in the Ulva mitogenomes. The three cfORFs including orf541/511, orf316 and orf227 were present in all sequenced Ulva mitogenomes, but their function is unknown based on the BLASTP searches in public databases of NCBI.
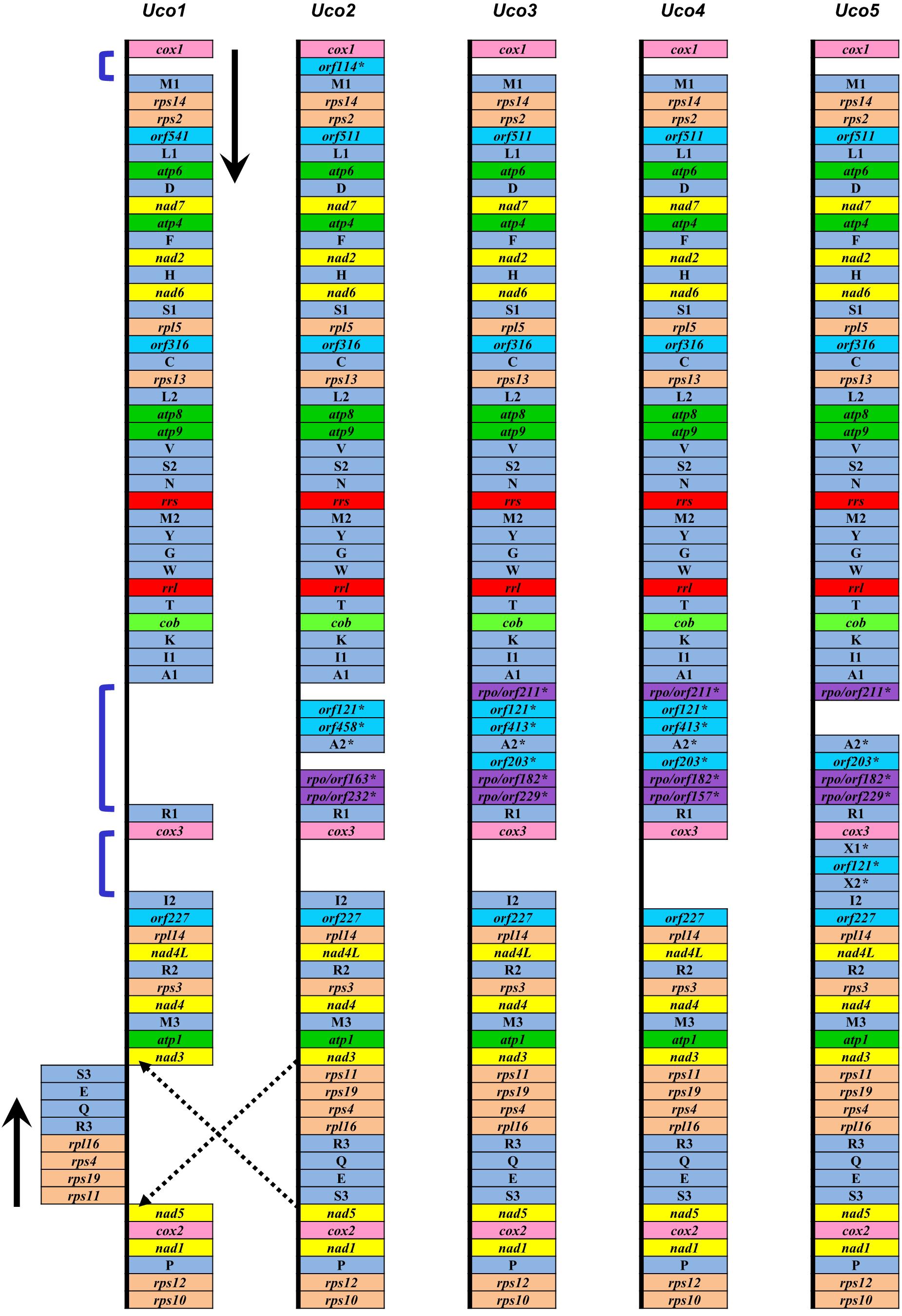
Figure 3. Comparison of genome organization and gene order of five U. compressa mitogenomes. The dark blue brackets showed three intergenic regions with the genome-specific tRNAs and orfs, which were indicated by an asterisk (∗).

Table 3. Comparison of insertion site, size and group of introns in the 17 Ulva mitogenomes.
The trnI(uau) between cox3 and rpl14 was absent in Uco4, but was present in the other four U. compressa mitogenomes (Figure 3), suggesting that this tRNA gene was subject to a recent loss or transfer in Uco4. Previously, we found that its counterpart was missing from two U. australis mitogenomes (Liu et al., 2017), but present in the other sequenced Ulva mitogenomes. This evidence indicates that trnI(uau) experienced at least two independent losses in the Ulva lineage. Interestingly, this tRNA gene displayed a conserved structure in DHU and TΨC stem-loops, but exhibited variable nucleotide sequences in the anticodon stem-loop. Instead of trnI with the anticodon TAT in U. compressa, it was predicted as trnK with TTT in U. australis, U. flexuosa and U. ohnoi, trnP with TGG in U. prolifera and U. linza, trnV with TAC in U. expansa, and trnX with TTTT in Ulva sp. (Figure 4). Although unusual, such a variation or modification of tRNA genes has been observed before in various organisms. For example, the tRNA gene located between nad4 and nad5 in brown algae displayed a variable anticodon among different species, which might play a new role instead of old function (Liu and Pang, 2015).
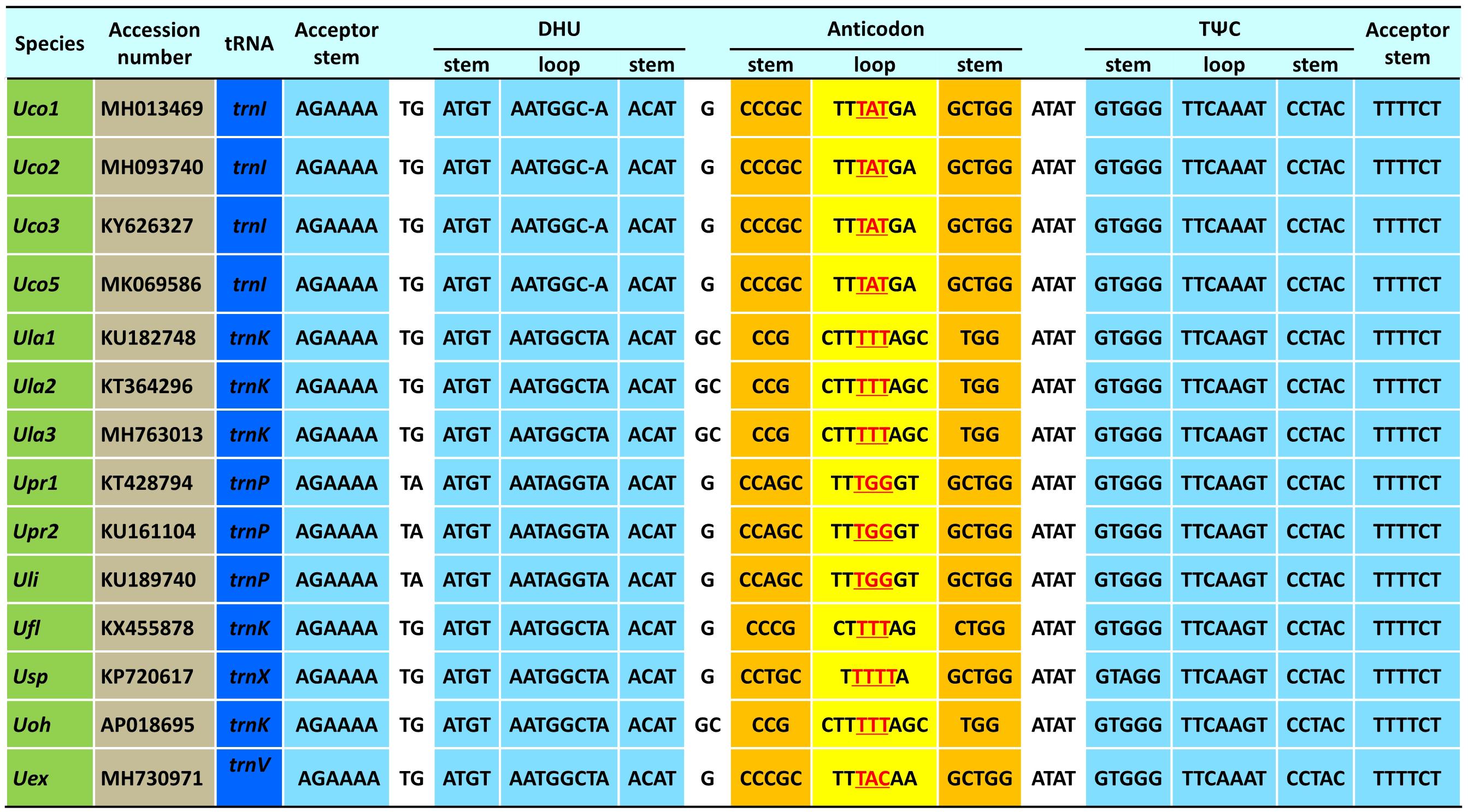
Figure 4. The aligned sequences of tRNA genes located between cox3 and rpl14 in the 14 Ulva mitogenomes.
Among the five U. compressa mitogenomes, a few genome-specific tRNAs and free-standing orfs (more than 100 codons) were observed in three intergenic regions, e.g., cox1-trnM1, trnA1-trnR1, and cox3-trnI2. The orf114 with unknown function was located in the intergenic region of cox1-trnM1 only in Uco2, while a DNA fragment carrying two pseudo tRNA genes (trnX1 and trnX2) and orf121 was present in the intergenic region of cox3-trnI2 only in Uco5 (Figure 3). Considering these two DNA fragments were absent in other Ulva mitogenome, it makes sense that two independent integration events of foreign DNA fragments happened in Uco2 and Uco5, respectively. Mitochondrial genomes have been observed to be able to accept foreign DNA fragments, which usually carry some specific tRNA genes and/or orfs, from diverse sources including chloroplast and nuclear sequences, bacteria, plasmid, virus, and jumping DNA/RNA-protein complex (e.g., Seif et al., 2005; Wang et al., 2007; Darracq et al., 2011; Apitz et al., 2013; Gandini and Sanchez-Puerta, 2017). However, the source of these foreign fragments observed in mitogenomes of U. compressa is still unknown due to little information on the possible function of these orfs. The insertion of large foreign DNA sequences could increase the size of mitochondrial genomes. The mitogenomes of Silene vulgaris (angiosperms) ranged in size from 361 to 429 kb and the unprecedented amounts of intraspecific diversity were due to frequent acquisition and rapid turnover of foreign sequences (Sloan et al., 2012).
It is worth noting that four samples from China (Uco2–5) shared a specific DNA fragment, which harbored 4–6 orfs and one tRNA gene (trnA2), in the intergenic region of trnA1-trnR1. Of these sfORFs, three have sequence similarity to the DNA-directed RNA polymerase (rpo), while the others had little sequence similarity to any PCGs in the GenBank database (Figure 3). Based on the comparative sequence analysis, it was shown that Uco3 has a more complete DNA fragment in this region, which contains a collinear block of seven genes (orf211-orf121-orf413-trnA2-orf203-orf182-orf229) compared with Uco2, Uco4 and Uco5. Subsequent insertion/deletion events have occurred in Uco2 and 4, which caused frameshift mutations and even gene loss in this region. However, this DNA fragment was absent in the United States sample Uco1 as well as the mitogenomes of the other Ulva species, indicating that the acquisition of this foreign fragment occurred in the common progenitor of Uco2-5 after its divergence from Uco1, as was consistent with the result of phylogenomic analysis (see a section below).
Variable Intron Content and Intronic orfs in U. compressa Mitogenomes
The U. compressa mitogenomes contained different intron content ranging from 4 introns in Uco3 and Uco4 to 7 in Uco1 (Table 3). These introns totally occupied 11.66% of the mitogenome in Uco2 to 20.24% in Uco1 (Table 2), suggesting that variation in intron content greatly caused changes in mitogenome size at the intraspecific level in U. compressa. Considering the previous similar findings in U. australis (Liu et al., 2017) and U. linza-prolifera (LP clade) (Liu and Pang, 2016; Zhou et al., 2016a, b), intraspecific variation of intron content probably occurs widely in Ulva species. Nine distinct insertion sites in four genes (cox1, cox2, nad3 and rnl) were detected among these five U. compressa mitogenomes. Comparative analysis of intron insertion-sites in U. compressa mitogenomes with that in other Ulva species indicated that cox1 tended to contain more introns, followed by rnl and rns, which was similar to that observed in other green algae, plants, Ochrophyta and fungi (e.g., Joardar et al., 2012; Sloan et al., 2012; Liu F. et al., 2020). However, group I introns prevailed in cox1 and rnl in Ulva species, as was significantly different from that observed in Ochrophyta (e.g., diatom) which harbored almost all group II introns (Liu F. et al., 2020).
Each intron in these five U. compressa mitogenomes contained one intron-encoded protein (orf with over 100 codons), which was believed to participate in intron propagation and/or intron splicing (maturases) (Dai et al., 2003; Haugen et al., 2005). All of the group I introns in U. compressa mitogenomes belonged to group IB and harbored an intronic orf which encoded a putative LAGLIDADG homing endonuclease (LHE), while all of the orfs in group IIA and IIB introns encoded putative reverse transcriptase/maturases (RTMs) in U. compressa. We found that these RTMs encoded by group IIA and IIB introns in Ulva species fall into two different lineages, group IIA and group IIB (Figure 5), which were observed to coevolve with the group II intron RNA structure (ribozyme components) (Fontaine et al., 1997; Toor et al., 2001; Zimmerly et al., 2001). However, a few of group II introns (i.e., intron cox1-874, rnl-2080, rnl-2698, rns-420 and rns-670) which harbored the homing endonucleases assigned to LAGLIDADG family (Liu et al., 2017), and a group ID intron (i.e., intron cox1-709) which contained a putative LHE, were absent in these five mitogenomes of U. compressa (Table 3).
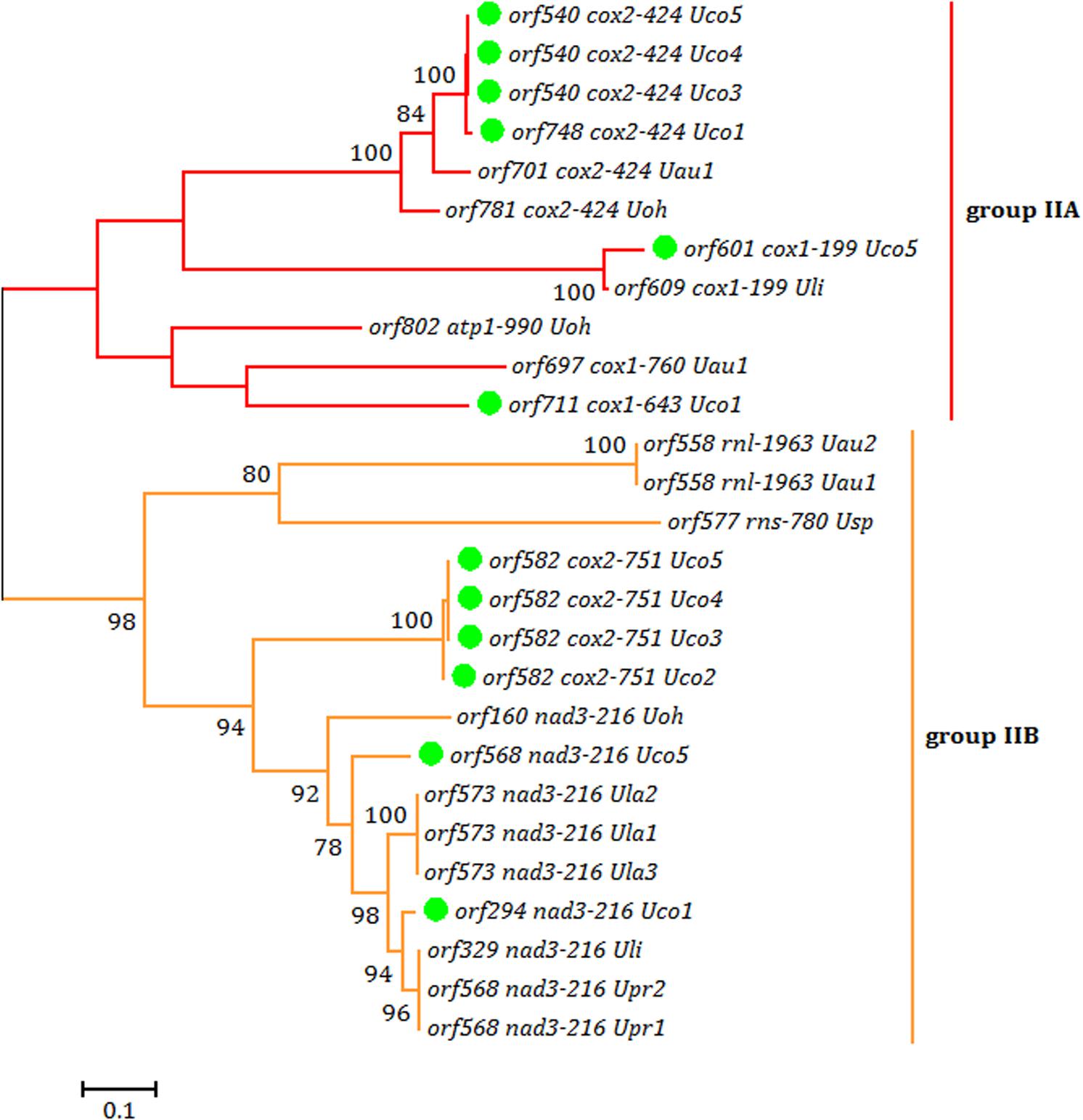
Figure 5. Phylogenetic tree based on Maximum Likelihood (ML) analysis of amino acid (aa) sequences of 27 reverse transcriptase (RT) domains from intronic orfs in group IIA and IIB introns. The ML analysis was conducted with 1,000 bootstrap replicates using MEGA 7.0. Branch lengths were proportional to the amount of sequence change, which were indicated by the scale bar below the trees.
It is notable that none of the introns was shared by all of the sequenced Ulva mitogenomes, suggesting that both group I and II introns were mobile genetic elements transferred across species (Dai et al., 2003; Haugen et al., 2005) and had the ability to invade specific target sites within some genes (Lang et al., 2007). Only two group IB introns in cox1 (i.e. intron cox1-1107 and cox1-1125) were shared by these five U. compressa mitogenomes (Table 3). These two group IB introns were common in most of Ulva species as well as species in Ulotrichales (Turmel et al., 2016). It seems more reasonable that these two group IB introns were inherited vertically from a common ancestor in the Ulvales/Ulotrichales lineage, and had experienced at least one or two independent losses in the Ulva lineage. Two group II introns in cox2 (i.e. intron cox2-424 and cox2-751) were present in four out of five U. compressa mitogenomes, but they belong to group IIA and IIB introns, respectively. Although these two group II introns were also present in one or two other Ulva species, they were more likely to be from multiple horizontal transfers to these specific insertion sites in the Ulva lineage (Turmel et al., 2016; Liu et al., 2017). Remarkably, intron cox1-643 (IIA) which contained a complete RTM protein was specific to Uco1, and absent in Uco2-5 and other Ulva species, indicating this group IIA intron should be the result of a recent independent invasion event.
Both group I and II introns from the same insertion site were homologous among the Ulva mitogenomes, indicating that cognate introns should have descended from a common progenitor (Ikuta et al., 2008). For the cognate introns of both group I and II, the RNA secondary structures of ribozyme components were highly conserved, while the intronic orf (LHE or RTM) might have been degenerated and even completely lost in some introns (Table 3). We observed that a total of 20 introns (~16.5%) in the 17 mitogenomes displayed degenerate orfs or no informative orf due to deletion/insertion and frameshift mutations. The U. expansa mitogenome harbored more degenerate introns than the other Ulva mitogenomes, of which some (e.g., intron rnl-2747, rns-420, and rns-670) completely lost the intronic orfs (i.e., LHEs). These degenerate introns have probably lost mobility competence, but they must retain splicing competence because they are located in conserved genes (Robart and Zimmerly, 2005).
Repeat Sequences and Intraspecific Genome Rearrangement
Numerous repeat sequences including tandem repeats (TRs) and inverted repeats (IRs) were distributed in the five mitogenomes of U. compressa. Most of these repeat sequences were restricted to the intergenic or intronic non-coding regions. The AT-rich TR, which was previously identified in the nad6-trnS(gcu) intergenic region among Ulva mitogenomes (Liu and Pang, 2016; Liu et al., 2017), showed differences in periodic sequence and copy number not only at interspecific level, but also at intraspecific level. This TR exhibited four types of periodic sequence × copy number in U. compressa, e.g. 16 bp × 5 in Uco1, 14 bp × 16.7 in Uco2, 4 bp × 71.8 in Uco3 and 4, and 44 bp × 4.4 in Uco5. Comparative analysis indicated that U. compressa mitogenomes contained many more IRs, ranging from 45 in Uco1 to 88 in Uco2, than that of the other Ulva species (3 - 34).
All of the genes annotated in Uco2-5 were coded on the same strand and shared a high level of conservation in gene synteny, as was the same as that in the other Ulva mitogenomes (Melton et al., 2015; Liu et al., 2017), but was different from the mitogenomes of Ulotrichales, Bryopsidales and Oltmannsiellopsidales which had their genes distributed on the two strands (Pombert et al., 2006; Turmel et al., 2016; Repetti et al., 2020). However, a locally collinear block of eight genes (rps11-rps19-rps4-rpl16-trnR-trnQ-trnE-trnS) with the size of 3,631 bp has been inverted only in Uco1 (Figure 6), indicating that this rearrangement event happened after its divergence from Uco2-5.
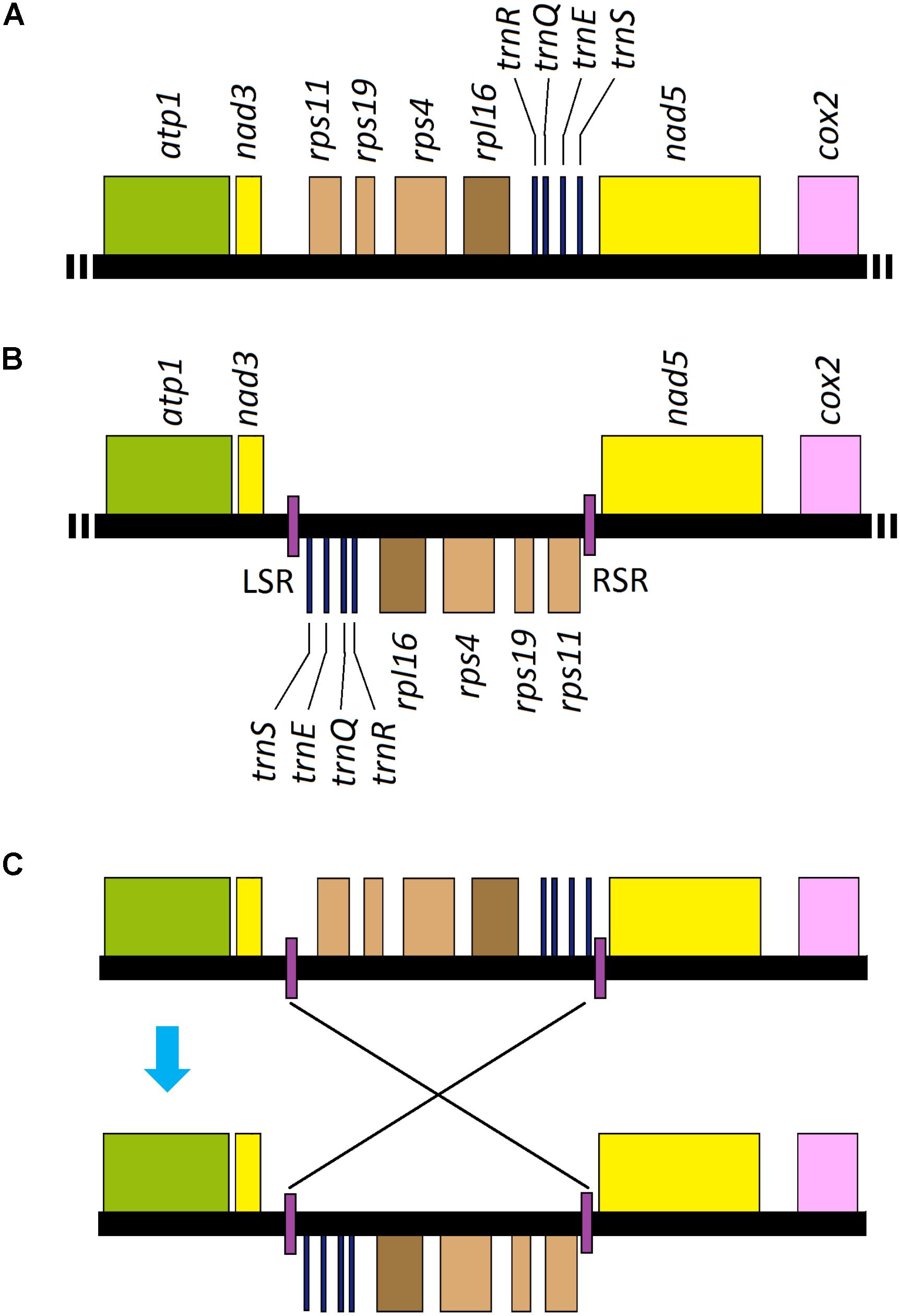
Figure 6. Intraspecific genome rearrangement of U. compressa mitogenome: (A) gene order from nad3 to nad5 in mitogenomes of Uco2-5 as well as the other Ulva species; (B) gene order from nad3 to nad5 in mitogenome of Uco1 with the specific inverted repeat (IR), and its left stem region (LSR) and right stem region (RSR) were located in two boundaries of the inverted eight gene block, respectively, and (C) hypothesis of the rearrangement event related to the specific IR element in Uco1.
Interestingly, a GC-rich (61.26%) IR element was located in the boundary of this inverted gene block in Uco1. The copies of this specific IR (left vs right: 150 vs 152 bp) differed by 1.97% mismatches and 1.32% indels, and its loop region was 3.7 kb in size carrying the eight genes (Figure 6). The absence of this IR from mitogenomes of Uco2-5 as well as the other Ulva mitogenomes not only eliminated the possibility that the IR was lost from the mitogenomes of Uco2-5, but also indicated that the IR appeared in Uco1 after its divergence from Uco2-5. IRs are common elements widespread in most of organelle genomes as well as the genomes of eukaryotes and prokaryotes. These elements could mediate genome rearrangement through excision of the repeat associated regions (Bi and Liu, 1996). IR sequences capable of forming stem-loop structures are the substrates for intragenomic recombination (Lin et al., 2001; Alverson et al., 2011). The mitogenomes of animals, fungi, plants and green algae are thought to undergo genome rearrangements mediated by IRs or stem-and-loop structures. The rearrangement event observed in Uco1 was probably related to the appearance of this specific IR element.
Intraspecific Gene Mutations and Phylogenomic Analysis
The majority of the common genes (76%) including 24 core PCGs, 2 rRNAs, 26tRNAs and 2 cfORFs displayed high identity values (> 98%) among these five mitogenomes of U. compressa (Supplementary Table S3), while some low values were observed in six genes (nad3, rps2, rps3, rps4, rps14 and orf541/511) mainly due to duplication mutations and insertion/deletion mutations of small DNA fragments at the intraspecific level. A 15-bp deletion mutation occurred at position 217 of nad3 in Uco1, not in Uco2-5. Interestingly, an identical sequence with a 27-bp hairpin structure was inserted three times into rps2 of Uco1 (position 756, 1662 and 1920), and twice into rps3 of Uco1 (position 375 and 406), and was not detected in the other four mitogenomes of U. compressa. The rps4 in Uco4 was shorter due to a C-insertion mutation at position 447 which caused the early appearance of stop codon. A duplication mutation of GCCCAAAGGCAAGGG happened at position 452 of rps14 in Uco1 and 3-5, but not in Uco2. Two insertion sequences with 51-bp and 39-bp hairpin structures were observed at position 587 and 677 of orf541 only in Uco1, which caused it to be longer than its counterparts (orf511) in the other four mitogenomes of U. compressa.
Phylogenomic analysis revealed that five samples of U. compressa formed a highly supported U. compressa clade (100% bootstrap) (Figure 7). Two samples from China (Uco3 and Uco5) closely grouped together with Uco4 from Southern Yellow Sea China, forming a subclade representing the filamentous, highly branched morphotype. This morphotype is common in Subei Shoal, Jiangsu, China and has partly contributed to the annual large-scale green tides in the Yellow Sea (Liu et al., 2013; Cai et al., 2018a). These three samples clustered more closely with Uco2 from Qingdao, Shandong, China than Uco1 from Swansboro, North Carolina, United States. The former is a ribbon, blade-like morphotype which was common in Qingdao coasts, China, but not found in the Subei Shoal, while the latter is a foliated, free-floating morphotype from the USA. The phylogenetic relationships among these five mitogenomes were consistent with the observed genome variations including integration of foreign DNA fragments, genome rearrangement, and gene mutations.
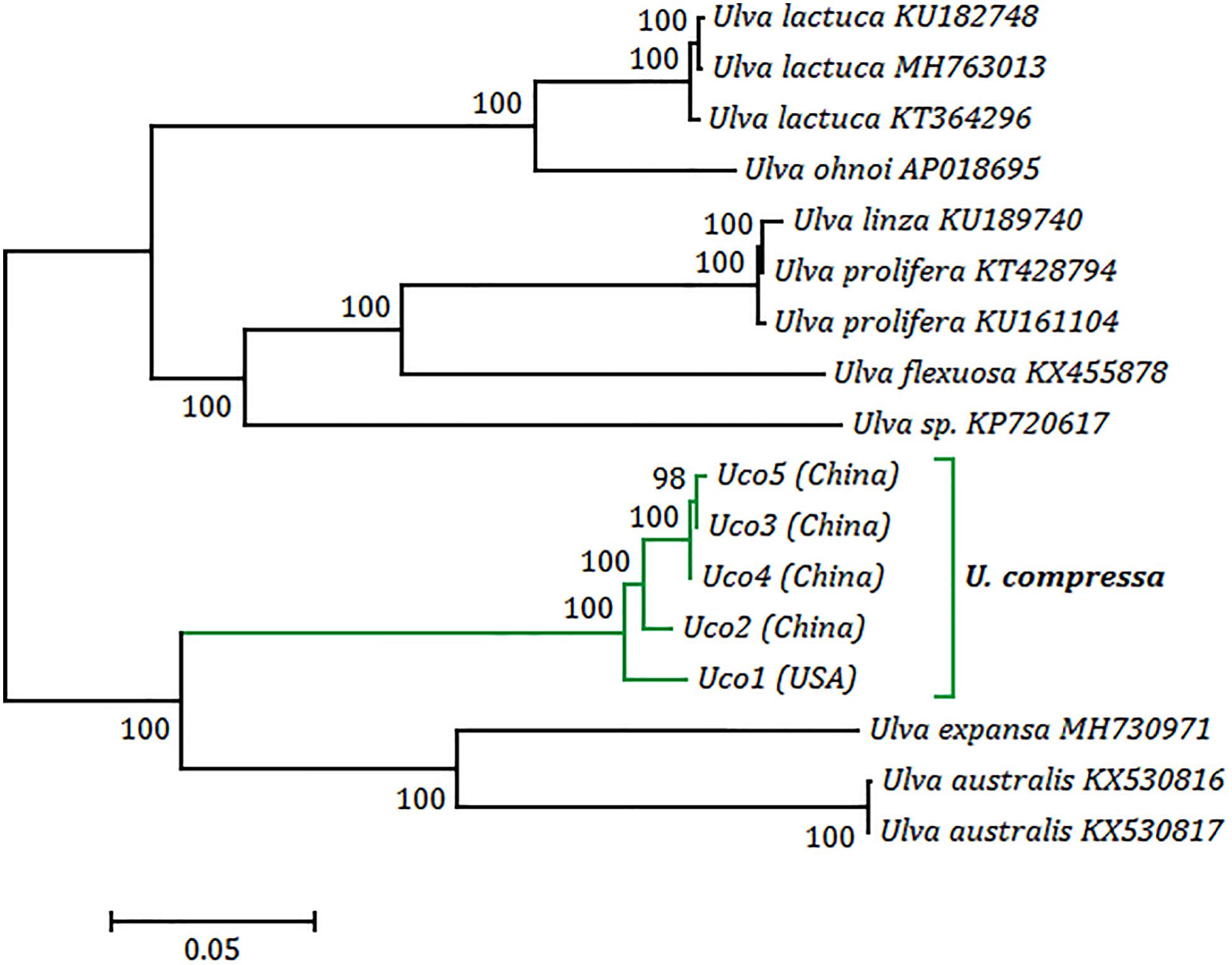
Figure 7. Phylogenomic tree based on Maximum Likelihood (ML) analysis of nucleotide sequences of 17 Ulva mitogenomes. The ML analysis was conducted with 1,000 bootstrap replicates using MEGA 7.0. Branch lengths were proportional to the amount of sequence change, which were indicated by the scale bar below the trees.
Although these samples of U. compressa exhibited diverse morphotypes, we found that they shared highly identical sequences of mitochondrial genes and other barcoding markers (ITS and rbcL). Based on a comparative analysis of the aligned nucleotide sequences of 61 mitochondrial genes included 29 core PCGs, 2 rRNAs, 27 tRNAs and 3 cfORFs, the sequence identity values ranged from 98.2% (different in 620 bp) in a pair of Uco1/4 to 99.9% in Uco3/5 (different in 19 bp) among these five U. compressa samples (Table 4). It is worth noting that based on comparative mitogenome information among these five samples of U. compressa, new DNA markers could be designed to study the population diversity and phylogeography of the green-tide forming alga (Steinhagen et al., 2019). Considering the marked intraspecific variations in genome size, intron content, genome architecture and gene mutations in U. compressa, the mitogenome will be a valuable tool to help us understand the native or non-indigenous nature of the cosmopolitan Ulva species.
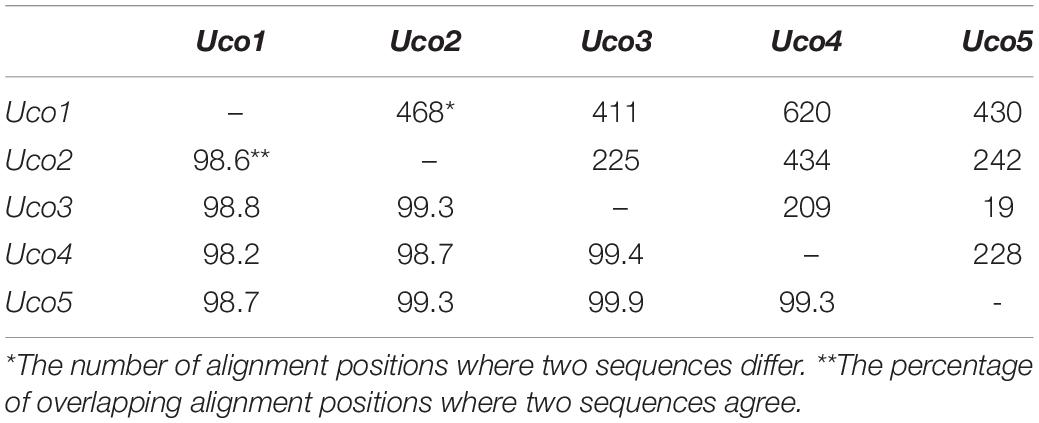
Table 4. Differences (bp, upper-right) and identity values (%, bottom-left) of the aligned nucleotide sequences of 61 genes included 29 core PCGs, 2 rRNAs, 27 tRNAs and 3 cfORFs in the five mitogenomes of U. compressa.
Conclusion
Although the sequenced mitogenomes from nine Ulva species showed some conserved features in core gene content and gene order, we still know little on the evolution of Ulva mitochondrial genomes at the intraspecific level. In this study, the mitogenomes of U. compressa displayed marked multiple intraspecific variations in genome size, gene content, intron content and genome architecture. This was mainly due to insertion of foreign DNA fragments, frequent gain or loss of both group I and II introns, enrichment of repeat sequences, and rearrangement by inversion of a syntenic block with eight collinear genes. Our study not only presented the first case of multiple intraspecific variations in ulvophycean mitogenomes, but also indicated that the mitogenome will be a valuable tool for understanding the native or non-indigenous nature of the cosmopolitan Ulva species. These results will enhance our understanding of the evolution of mitochondrial genomes in green algae.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/ Supplementary Material.
Author Contributions
FL and JM designed the study. FL, JM, and JL-B performed the experiments. FL, JM, and NC performed the analysis. FL wrote the manuscript. All authors have read and approved the final version of the manuscript.
Funding
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (Nos. XDA23050302 and XDA23050403 to FL), the National Natural Science Foundation of China (No. 41876165 to FL), the Key Research Program of Frontier Sciences, Chinese Academy of Sciences (No. QYZDB-SSW-DQC023 to FL), the Major Scientific and Technological Innovation Project of Shandong Province (No. 2019JZZY020706 to FL), the Director Fund of Pilot National Laboratory for Marine Science and Technology (Qingdao) (No. QNLM201704), the CAS Pioneer Hundred Talents Program (to NC), the Taishan Scholar Project Special Fund (to NC), and the National Science Foundation (NSF) HBCU-UP (No. 1436759) for postdoctoral fellowship (to JM).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We wish to thank Wei Luan, Yu Wang, and Zhe Jin for their assistance in algal collection and data analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.00714/full#supplementary-material
Footnotes
- ^ Biomatters, http://www.geneious.com.
- ^ http://www.ncbi.nlm.nih.gov/gorf/orFigurecgi
- ^ http://tandem.bu.edu/cgi-bin/irdb/irdb.exe
References
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Alverson, A. J., Rice, D. W., Dickinson, S., Barry, K., and Palmer, J. D. (2011). Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber. Plant Cell 23, 2499–2513. doi: 10.1105/tpc.111.087189
Apitz, J., Weihe, A., Pohlheim, F., and Börner, T. (2013). Biparental inheritance of organelles in Pelargonium: evidence for intergenomic recombination of mitochondrial DNA. Planta 237, 509–515. doi: 10.1007/s00425-012-1768-x
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bi, X., and Liu, L. F. (1996). DNA rearrangement mediated by inverted repeats. Proc. Natl. Acad. Sci. U S A. 93, 819–823. doi: 10.1073/pnas.93.2.819
Blomster, J., Bäck, S., Fewer, D. P., Kiirikki, M., Lehvo, A., Maggs, C. A., et al. (2002). Novel morphology in Enteromorpha (Ulvophyceae) forming green tides. Am. J. Bot. 89, 1756–1763. doi: 10.3732/ajb.89.11.1756
Burger, G., and Nedelcu, A. M. (2012). “Mitochondrial genomes of algae,” in Advances in Photosynthesis and Respiration Including Bioenergy and Related Processes: Genomics of Chloroplasts and Mitochondria, eds R. Bock and V. Knoop, (Dordrecht: Springer), 125–157.
Cai, C., Liu, F., Jiang, T., Wang, L., Jia, R., Zhou, L., et al. (2018a). Comparative study on mitogenomes of green tide algae. Genetica 146, 529–540. doi: 10.1007/s10709-018-0046-7
Cai, C., Wang, L., Jiang, T., Zhou, L., He, P., and Jiao, B. (2018b). The complete mitochondrial genomes of green tide algae Ulva flexuosa (Ulvophyceae. Chlorophyta). Conserv. Genet. Resour. 10, 415–418. doi: 10.1007/s12686-017-0838-6
Cocquyt, E., Verbruggen, H., Leliaert, F., and De Clerck, O. (2010). Evolution and cytological diversification of the green seaweeds (Ulvophyceae). Mol. Biol. Evol. 27, 2052–2061. doi: 10.1093/molbev/msq091
Dai, L., Toor, N., Olson, R., Keeping, A., and Zimmerly, S. (2003). Database for mobile group II introns. Nucleic Acids Res. 31, 424–426. doi: 10.1093/nar/gkg049
Darracq, A., Varre, J. S., Marechal-Drouard, L., Courseaux, A., Castric, V., Saumitou-Laprade, P., et al. (2011). Structural and content diversity of mitochondrial genome in beet: a comparative genomic analysis. Genome Biol. Evol. 3, 723–736. doi: 10.1093/gbe/evr042
Fontaine, J. M., Goux, D., Kloareg, B., and Loiseaux-de Goër, S. (1997). The reverse-transcriptase-like proteins encoded by group II introns in the mitochondrial genome of the brown alga Pylaiella littoralis belong to two different lineages which apparently coevolved with the group II ribosyme lineages. J. Mol. Evol. 44, 33–42. doi: 10.1007/pl00006119
Fuèíková, K., Leliaert, F., Cooper, E. D., Škaloud, P., D’Hondt, S., De Clerck, O., et al. (2014). New phylogenetic hypotheses for the core Chlorophyta based on chloroplast sequence data. Front. Ecol. Evol. 2:63. doi: 10.3389/fevo.2014.00063
Gandini, C. L., and Sanchez-Puerta, M. V. (2017). Foreign plastid sequences in plant mitochondria are frequently acquired via mitochondrion-to-mitochondrion horizontal transfer. Sci. Rep. 7:43402.
Gray, M. W., Burger, G., and Lang, B. F. (2001). The origin and early evolution of mitochondria. Genome Biol. 2, reviews1018.1–reviews1018.5.
Guiry, M. D., and Guiry, G. M. (2020). AlgaeBase. World-wide Electronic Publication, Galway: National University of Ireland
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucl. Acids Symp. Ser. 41, 95–98.
Hanyuda, T., Heesch, S., Nelson, W., Sutherland, J., Arai, S., Boo, S. M., et al. (2016). Genetic diversity and biogeography of native and introduced populations of Ulva pertusa (Ulvales. Chlorophyta). Phycol. Res. 64, 102–109. doi: 10.1111/pre.12123
Haugen, P., Simon, D. M., and Bhattacharya, D. (2005). The natural history of group I introns. Trends Genet. 21, 111–119. doi: 10.1016/j.tig.2004.12.007
Hayden, H. S., and Waaland, J. R. (2002). Phylogenetic systematics of the Ulvaceae (Ulvales. Ulvophyceae) using chloroplast and nuclear DNA sequences. J. Phycol. 38, 1200–1212. doi: 10.1046/j.1529-8817.2002.01167.x
Hughey, J. R., Maggs, C. A., Mineur, F., Jarvis, C., Miller, K. A., Shabaka, S. H., et al. (2019). Genetic analysis of the Linnaean Ulva lactuca (Ulvales. Chlorophyta) holotype and related type specimens reveals name misapplications, unexpected origins, and new synonymies. J. Phycol. 55, 503–508. doi: 10.1111/jpy.12860
Hughey, J. R., Miller, K. A., and Gabrielson, P. W. (2018). Mitogenome analysis of a green tide forming Ulva from California. USA confirms its identity as Ulva expansa (Ulvaceae, Chlorophyta). Mitochondrial DNA Part B. 3, 1302–1303. doi: 10.1080/23802359.2018.1535859
Ikuta, K., Kawai, H., Müller, D. G., and Ohama, T. (2008). Recurrent invasion of mitochondrial group II introns in specimens of Pylaiella littoralis (brown alga), collected worldwide. Curr Genet. 53, 207–216. doi: 10.1007/s00294-008-0178-x
Joardar, V., Abrams, N. F., Hostetler, J., Paukstelis, P. J., Pakala, S., Pakala, S. B., et al. (2012). Sequencing of mitochondrial genomes of nine Aspergillus and Penicillium species identifies mobile introns and accessory genes as main sources of genome size variability. BMC Genom. 13:698. doi: 10.1186/1471-2164-13-698
Jones, D. T., Taylor, W. R., and Thornton, J. M. (1992). The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282. doi: 10.1093/bioinformatics/8.3.275
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lang, B. F., Laforest, M.-J., and Burger, G. (2007). Mitochondrial introns: a critical view. Trends Genet. 23, 119–125. doi: 10.1016/j.tig.2007.01.006
Leliaert, F., Smith, D. R., Moreau, H., Herron, M. D., Verbruggen, H., Delwiche, C. F., et al. (2012). Phylogeny and molecular evolution of the green algae. Crit. Rev. Plant Sci. 31, 1–46.
Lin, C. T., Lin, W. H., Lyu, Y. L., and Whang-Peng, J. (2001). Inverted repeats as genetic elements for promoting DNA inverted duplication: implications in gene amplification. Nucleic Acids Res. 29, 3529–3538. doi: 10.1093/nar/29.17.3529
Liu, F., and Pang, S. J. (2015). Mitochondrial genome of Turbinaria ornata (Sargassaceae. Phaeophyceae): comparative mitogenomics of brown algae. Curr. Genet. 61, 621–631. doi: 10.1007/s00294-015-0488-8
Liu, F., and Pang, S. (2016). The mitochondrial genome of the bloom-forming green alga Ulva prolifera. Mitochondrial DNA Part A. 27, 4530–4531. doi: 10.3109/19401736.2015.1101548
Liu, F., Liu, S., Huang, T., and Chen, N. (2020). Construction and comparative analysis of mitochondrial genome in the brown tide forming alga Aureococcus anophagefferens (Pelagophyceae. Ochrophyta). J. Appl. Phycol. 32, 441–450. doi: 10.1007/s10811-019-01952-0
Liu, F., Melton, J. T., and Bi, Y. (2017). Mitochondrial genomes of the green macroalga Ulva pertusa (Ulvophyceae. Chlorophyta): novel insights into the evolution of mitogenomes in the Ulvophyceae. J. Phycol. 53, 1010–1019. doi: 10.1111/jpy.12561
Liu, F., Pang, S., Chopin, T., Gao, S., Shan, T., Zhao, X., et al. (2013). Understanding the recurrent large-scale green tide in the Yellow Sea: temporal and spatial correlations between multiple geographical, aquacultural and biological factors. Mar. Environ. Res. 83, 38–47. doi: 10.1016/j.marenvres.2012.10.007
Liu, F., Pang, S., Chopin, T., Xu, N., Shan, T., Gao, S., et al. (2010). The dominant Ulva strain of the 2008 green algal bloom in the Yellow Sea was not detected in the coastal waters of Qingdao in the following winter. J. Appl. Phycol. 22, 531–540. doi: 10.1007/s10811-009-9489-7
Liu, M., Liu, F., Chen, N., Melton, J. T., and Luo, M. (2020). Mitochondrial genomes and phylogenomic analysis of Ulva lactuca Linnaeus (Ulvophyceae. Chlorophyta). Mitochondrial DNA Part B. 5, 1638–1639. doi: 10.1080/23802359.2020.1745712
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1:18.
Melton, J. T., Leliaert, F., Tronholm, A., and Lopez-Bautista, J. M. (2015). The complete chloroplast and mitochondrial genomes of the green macroalga Ulva sp. UNA00071828 (Ulvophyceae, Chlorophyta). PLoS One 10:e0121020. doi: 10.1371/journal.pone.0121020
Melton, J. T., and Lopez-Bautista, J. M. (2016). De novo assembly of the mitochondrial genome of Ulva fasciata Delile (Ulvophyceae. Chlorophyta), a distromatic blade-forming green macroalga. Mitochondrial DNA Part A. 27, 3817–3819. doi: 10.3109/19401736.2015.1082095
Mine, I., Menzel, D., and Okuda, K. (2008). Morphogenesis in giant-celled algae. Int. Rev. Cell Mol. Biol. 266, 37–83. doi: 10.1016/s1937-6448(07)66002-x
Pombert, J.-F., Beauchamp, P., Otis, C., Lemieux, C., and Turmel, M. (2006). The complete mitochondrial DNA sequence of the green alga Oltmannsiellopsis viridis: evolutionary trends of the mitochondrial genome in the Ulvophyceae. Curr. Genet. 50, 137–147. doi: 10.1007/s00294-006-0076-z
Pombert, J.-F., Otis, C., Lemieux, C., and Turmel, M. (2004). The complete mitochondrial DNA sequence of the green alga Pseudendoclonium akinetum (Ulvophyceae) highlights distinctive evolutionary trends in the Chlorophyta and suggests a sister-group relationship between the Ulvophyceae and Chlorophyceae. Mol. Biol. Evol. 21, 922–935. doi: 10.1093/molbev/msh099
Punta, M., Coggill, P. C., and Eberhardt, R. Y. (2012). The Pfam protein families database. Nucleic Acids Res. 40, D290–D301.
Repetti, S. I., Jackson, C. J., Judd, L. M., Wick, R. R., Holt, K. E., and Verbruggen, H. (2020). The inflated mitochondrial genomes of siphonous green algae reflect processes driving expansion of noncoding DNA and proliferation of introns. Peer J. 8:e8273. doi: 10.7717/peerj.8273
Robart, A. R., and Zimmerly, S. (2005). Group II intron retroelements: function and diversity. Cytogenet Genome Res. 110, 589–597. doi: 10.1159/000084992
Sauvage, T., Schmidt, W. E., Yoon, H. S., Paul, V. J., and Fredericq, S. (2019). Promising prospects of nanopore sequencing for algal hologenomics and structural variation discovery. BMC Genom. 20:850. doi: 10.1186/s12864-019-6248-2
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, 686–689.
Seif, E., Leigh, J., Liu, Y., Roewer, I., Forget, L., and Lang, B. F. (2005). Comparative mitochondrial genomics in zygomycetes: bacteria-like RNase P RNAs, mobile elements and a close source of the group I intron invasion in angiosperms. Nucleic Acids Res. 33, 734–744. doi: 10.1093/nar/gki199
Sloan, D. B., Müller, K., McCauley, D. E., Taylor, D. R., and Štorchová, H. (2012). Intraspecific variation in mitochondrial genome sequence, structure, and gene content in Silene vulgaris, an angiosperm with pervasive cytoplasmic male sterility. New Phytol. 196, 1228–1239. doi: 10.1111/j.1469-8137.2012.04340.x
Steinhagen, S., Weinberger, F., and Karez, R. (2019). Molecular analysis of Ulva compressa (Chlorophyta. Ulvales) reveals its morphological plasticity, distribution and potential invasiveness on German North Sea and Baltic Sea coasts. Eur. J. Phycol. 54, 102–114. doi: 10.1080/09670262.2018.1513167
Suzuki, S., Yamaguchi, H., Hiraoka, M., and Kawachi, M. (2018). Mitochondrial and chloroplast genome sequences of Ulva ohnoi, a green-tide forming macroalga in the Southern coastal regions of Japan. Mitochondrial DNA Part B. 3, 765–767. doi: 10.1080/23802359.2018.1483778
Tamura, K., and Nei, M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10, 512–526.
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., and Higgins, D. G. (1997). The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882. doi: 10.1093/nar/25.24.4876
Toor, N., Hausner, G., and Zimmerly, S. (2001). Coevolution of group II intron RNA structures with their intron-encoded reverse transcriptases. RNA 7, 1142–1152. doi: 10.1017/s1355838201010251
Tritt, A., Eisen, J. A., Facciotti, M. T., and Darling, A. E. (2012). An integrated pipeline for de novo assembly of microbial genomes. PLoS One 7:e42304. doi: 10.1371/journal.pone.0042304
Turmel, M., Otis, C., and Lemieux, C. (2016). Mitochondrion-to-chloroplast DNA transfers and intragenomic proliferation of chloroplast group II introns in Gloeotilopsis green algae (Ulotrichales. Ulvophyceae). Genome Biol. Evol. 8, 2789–2805. doi: 10.1093/gbe/evw190
Wang, D., Wu, Y. W., Shih, A. C. C., Wu, C. S., Wang, Y. N., and Chaw, S. M. (2007). Transfer of Chloroplast genomic DNA to mitochondrial genome occurred at least 300 MYA. Mol. Biol. Evol. 24, 2040–2048. doi: 10.1093/molbev/msm133
Wang, Y., Liu, F., Liu, X., Shi, S., Bi, Y., and Moejes, F. W. (2019). Comparative transcriptome analysis of four co-occurring Ulva species for understanding the dominance of Ulva prolifera in the Yellow Sea green tides. J. Appl. Phycol. 31, 3303–3316. doi: 10.1007/s10811-019-01810-z
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Zheng, F., Liu, H., Jiang, M., Xu, Z., Wang, Z., Wang, C., et al. (2018). The complete mitochondrial genome of the Caulerpa lentillifera (Ulvophyceae. Chlorophyta): Sequence, genome content, organization structure and phylogenetic consideration. Gene 673, 225–238. doi: 10.1016/j.gene.2018.06.050
Zhou, L., Wang, L., Zhang, J., Cai, C., and He, P. (2016a). Complete mitochondrial genome of Ulva linza, one of the causal species of green macroalgal blooms in Yellow Sea. China. Mitochondrial DNA Part B. 1, 31–33. doi: 10.1080/23802359.2015.1137806
Zhou, L., Wang, L., Zhang, J., Cai, C., and He, P. (2016b). Complete mitochondrial genome of Ulva prolifera, the dominant species of green macroalgal blooms in Yellow Sea. China. Mitochondrial DNA Part B. 1, 76–78. doi: 10.1080/23802359.2015.1137831
Zimmerly, S., Hausner, G., and Wu, X. (2001). Phylogenetic relationships among group II intron ORFs. Nucleic Acids Res. 29, 1238–1250. doi: 10.1093/nar/29.5.1238
Keywords: green algae, mitochondrial genome, intraspecific evolution, genome rearrangement, Ulvophyceae, intron
Citation: Liu F, Melton JT III, Lopez-Bautista JM and Chen N (2020) Multiple Intraspecific Variations of Mitochondrial Genomes in the Green-Tide Forming Alga, Ulva compressa Linnaeus (Ulvophyceae, Chlorophyta). Front. Mar. Sci. 7:714. doi: 10.3389/fmars.2020.00714
Received: 09 June 2020; Accepted: 05 August 2020;
Published: 27 August 2020.
Edited by:
Andrew Stanley Mount, Clemson University, United StatesReviewed by:
Yongyu Zhang, Qingdao Institute of Bioenergy and Bioprocess Technology (CAS), ChinaJian-Wen Qiu, Hong Kong Baptist University, Hong Kong
Copyright © 2020 Liu, Melton, Lopez-Bautista and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Liu, liufeng@qdio.ac.cn